Como citar
Cabrera Pérez J. Síndrome de Angelman reporte de un caso. ADOPA. 2023;1(1):47-58. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/4
ADOPA, Vol. I, No. 1, enero-abril, 2023 • ISSN (en línea): 2960-7582 • Sitio web: https://adopa.pediatriadominicana.org/index.php/adopa
Cabrera Pérez J. Síndrome de Angelman reporte de un caso. ADOPA. 2023;1(1):47-58. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/4
Introducción: las manifestaciones clínicas más sobresalientes del síndrome de Angelman comprenden un retraso severo en el desarrollo psicomotor, ausencia de lenguaje verbal, convulsiones frecuentes, expresión permanente de rostro feliz con una sonrisa no motivada y marcha tambaleante, y dismorfia craneofacial. Es un trastorno genético por deleción del cromosoma 15 (15q11q13) en su expresión materna. En alrededor de un 10 a 15 % de los pacientes las pruebas genéticas son normales, como ocurrió en nuestro paciente. Su prevalencia se estima en 1/15000-20000 en la población general.
Objetivo: dar a conocer la existencia del Síndrome de Angelman en República Dominicana a toda la comunidad médica, especialmente a neurólogos y pediatras, como una causa importante de retraso psicomotor severo y de convulsiones.
Materiales y método: se proporciona una paciente adolescente de 16 años con unas características fenotípicas de alteraciones genéticas y evidente retraso psicomotor. Cuya expresión clínica es compatible con el síndrome de Angelman.
Conclusiones: por la asociación de retraso severo mental con un fenotipo conductual característico de expresión de felicidad, se le llegó a conocer inapropiadamente como marioneta feliz, pero la existencia de convulsiones nos debe hacer pensar en el síndrome de Angelman.
Introduction: the most salient clinical manifestations of Angelman syndrome include a severe delay in psychomotor development, absence of verbal language, frequent seizures, permanent expression of happy face with an unmotivated smile and wobbly gait, and craniofacial dysmorphism. It is a genetic disorder due to deletion of chromosome 15 (15q11q13) in its maternal expression. In about 10 to 15 % of patients, genetic testing is normal, as it was in our patient. Its prevalence is estimated at 1/15000-20000 in the general population.
Objective: To make the existence of Angelman Syndrome known in our country to the entire medical community, especially neurologists and paediatricians. As an important cause of severe psychomotor retardation and seizures.
Materials and method: a 16-year-old adolescent patient with phenotypic characteristics of genetic alterations and evident psychomotor retardation is provided. Whose clinical expression is compatible with Angelman Syndrome.
Conclusions: the association of severe mental retardation with a characteristic behavioral phenotype of happiness expression, which is why it became inappropriately known as a happy puppet and the existence of seizures should make us think of Angelman syndrome.
El síndrome de Angelman (SA) es un trastorno neurogenético presente en aproximadamente 1/15,000 1/20,000 individuos. Se desconoce la incidencia exacta del SA, los estudios reflejan más la prevalencia que la incidencia.1-3 La primera descripción clínica la hizo el Dr. Harry Angelman en 1965, un pediatra inglés, de tres niños con retraso mental grave, ataxia, risa excesiva, convulsiones.4 SA es causado por expresión ausente del gen UBE3A de impronta materna.5-9 Las características incluyen retrasos en el desarrollo, especialmente en la expresividad del lenguaje, un comportamiento feliz distintivo, convulsiones, déficits motores gruesos y finos, temblores, trastornos del sueño, problemas gastrointestinales, conducta estereotipada, ansiedad, e hipercinesia.10, 11
En 1995 se establecieron por consenso los criterios de diagnóstico del síndrome de Angelman10 y una década después fueron revisados.11 Dichos criterios son válidos para los cuatros mecanismos genéticos que producen el síndrome de Angelman. Hay cuatro mecanismos moleculares reconocidos: (a) eliminación de 15q11.2q13 materno (70-75 %); (b) mutación del gen UBE3A materno (15 %) con un 50 % de recurrencia riesgo si se hereda por vía materna; raramente(c) disomía uniparental paterna (UPD, 5–7 %); y (d) defecto de impronta (Imp D, 5–7 %).
Hay un grupo de pacientes que no se encuentra ninguna base
genética.11
El recién nacido es un niño normal y alrededor del sexto mes puede observarse un retraso en el desarrollo psicomotor. La sintomatología típica no aparece hasta después del primer año.
Todos los pacientes presentan: historia prenatal y neonatal normal, perímetro craneal normal al nacer sin
defectos malformativos mayores. Estudios metabólicos, hematológicos y bioquímicos normales. No anomalías
estructurales del SNC en estudios de neuroimagen como TAC y resonancia magnética, aunque pueden observarse
ligeras atrofias corticales o desmielinización.
Se encuentran en 100 % de los casos:
Frecuente (más del 80 %)
Asociados (20-80 %)
Fecha: 31 de enero del 2011
Información proporcionada por: la madre.
Se trata de paciente femenina de 16 años, procedente de Inoa, San José de las Matas, hija de madre de 19 años G2, P2, A0, C0. Chequeos regulares, sin antecedentes mórbidos conocidos. Parto eutócico. Peso 9 ½ libras. Apgar: 7/9. Lactancia materna: 1 mes. Ablactación: 6 meses.
Sostén de la cabeza: 8 meses. Se sentó al 1 año. Gateó al 1 año.
Caminó: 2 años y 3 meses. Primera palabra: 5 años, monosílabo (MA). Control de esfínteres anal y vesical: 15 años. Resalta la madre que se siente atraída por el agua y disfruta mucho el verla en cualquier fuente.
Convulsiones, historia de constipación y retraso psicomotor.
Refiere informante, que desde el nacimiento la paciente presenta retraso en el desarrollo psicomotor manifestado por retraso para sostener la cabeza, sentarse y gatear, así como imposibilidad para articular palabras. A los tres años presentó movimientos tónicos-clónicos generalizados con desviación de la mirada hacia arriba, relajación de esfínteres anal y vesical en una ocasión de duración no especificada, motivo por el cual es llevada a centro de salud privado, donde es evaluada por neurólogo del centro. Se le realiza electroencefalograma y se determina que, por la clínica, además de los estudios realizados, la misma cursa con un retraso psicomotor, por lo que es manejada de forma ambulatoria con Valpakine por un año y luego de dicho período abandona el tratamiento.
Paciente persiste con alteración de su desarrollo psicomotor y a los 14 años de edad, nueva vez presenta
movimientos tónico-clónicos generalizados en dos ocasiones, de cinco minutos de duración aproximadamente,
acompañado de
salivación profusa (sialorrea), desviación de la mirada hacia arriba, sin relajación de esfínteres, motivo por
el cual es llevada a centro de salud público de su comunidad, donde es evaluada y manejada con diazepam y luego
referida a este centro, en el que es ingresada con diagnóstico de “Convulsiones de origen a investigar”, D/C
Epilepsia, D/C Tumor cerebral, Retraso psicomotor con manejo de solución mixta 0.33 %, difenilhidantoina y
medidas generales. Luego del ingreso se le realizaron electrolitos y Tac de cráneo cuyo reporte fue normal; la
paciente fue evaluada por el departamento de neurología, el cual consideró egresar con manejo de epival
(valproato) 500 mg cada 24 horas y seguimiento por consulta; después de dicho manejo se han controlado las
crisis convulsivas. En las figuras 2 y 3 se observan posiciones características de la paciente.
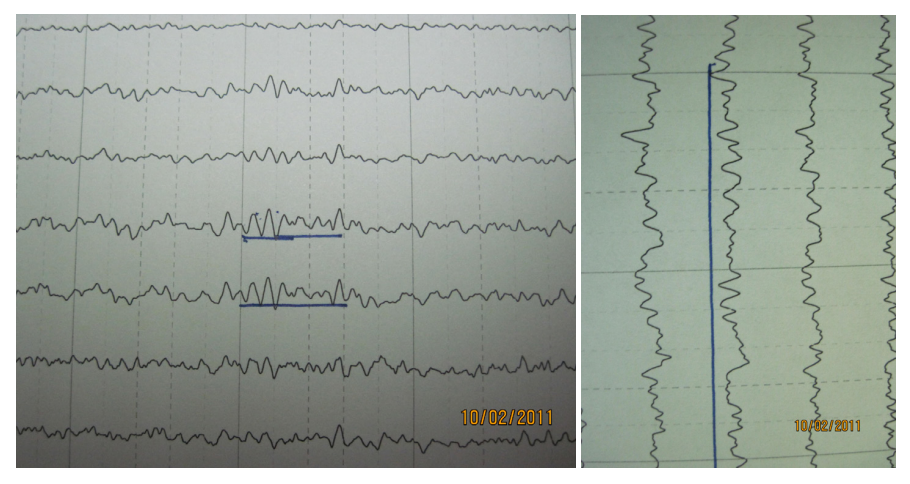
Figura 1. Electroencefalograma a su ingreso por la crisis convulsiva
Fue evaluada por departamento de Clínica antes de ser egresada. Al examen físico observamos una adolescente que
tenía una sonrisa constante, sin ninguna motivación, y estaba en un estado permanente de felicidad, con
movimientos gráciles de las manos que, involuntariamente, elevaba hacia arriba, abajo y a todos lados.
Presentaba al caminar una marcha tambaleante, con sacudidas del tronco hacia delante y hacia atrás, pausados y
constantes, que cesaban al poco tiempo para reiniciar nuevamente. Asimismo, evidenciaba movimientos de las
comisuras bucales masticatorios hacia todos lados y protrusión de la lengua sin ninguna coordinación ni
estímulo, marcha atáxica, se detenía por segundos, distraída, para iniciar nuevamente. Presentaba, además,
babeos frecuentes.

Figura 2a

Figura 2b
Figuras 2a. y 2 b Posición adoptada por la paciente
Fuente: Hospital Dr. Arturo Grullón.

Figura 2c

Figura 2d
Figuras 2c y 2d. Manos en posición de candelabro y sonrisa no motivada permanente. Dismorfismo facial
Fuente: Hospital Dr. Arturo Grullón.
Figura 3. Movimientos masticatorios
Fuente: Hospital Dr. Arturo Grullón.
El diagnóstico de síndrome de Angelman fue considerado. El diagnóstico diferencial incluye:
Se realizó una prueba genética de FISH, reportándose normal sin evidencia de deleciones cromosómicas.
El diagnóstico de la EA clásica suele ser hecho entre 1 y 2 años de edad, a menudo más tarde con presentación no clásica.3 Nuestro paciente tuvo un retraso en el desarrollo psicomotor severo desde su primer año de vida y debutó con una convulsión a los tres años, tal como se ha reportado en la literatura en los pacientes registrados con esta enfermedad, pero lamentablemente se tardó 16 años en hacerse el diagnóstico, a pesar de reunir los criterios clínicos para ser considerado el SA.
Creemos que el retraso severo en el desarrollo, la ausencia del lenguaje, la ataxia y los temblores y la conducta de felicidad con su sonrisa permanente, que está en el 100 % de los casos, así como en el 80 % de los casos dimorfismo facial, tal como se ha descrito, crisis convulsivas a partir de los 3 años de edad y el electroencefalograma de ondas de gran amplitud y polipuntas, las manos en posición prona y supina, en forma de marioneta o candelabro, el cuerpo hacia delante y hacia abajo, que denota una sacudida como se ve en figura 2, unida al lenguaje no verbal, que siempre tiene, y la marcha tambaleante y apraxia, no deja ninguna duda de que se trata de un SA. Posee además los hallazgos clínicos que se encuentran entre 20 y 80 % de los casos tales como problemas de succión, babeo frecuente, conducta masticatoria, marcha característica y fascinación por el agua.
No se realizaron otras pruebas genéticas, debido a la carencia de las mismas en esa época. Solo un en 15 %, aproximadamente, de los pacientes no se encuentra alteración genética. Creemos que reúne todos los requisitos para ser considerada como un de síndrome de Angelman.
Entre las limitaciones de nuestra presentación, está el no haberle realizado otras pruebas genéticas que pudieran
detectar una mutación del gen UBE3A y así poder decir con mayor nivel de certeza que se trata de un síndrome de
Angelman sin hallazgos genéticos. No podemos hacer comentarios sobre la evolución de la paciente, ya que
perdimos el contacto con esta por razones migratorias. El pronóstico es malo, pues, aunque tienen un promedio de
vida muy parecido a la población general, nunca logran mejorar la expresión verbal ni su retraso mental. Viven
con grandes limitaciones funcionales y muy pobre calidad de vida, tanto para los que padecen la enfermedad como
para los familiares, siendo una gran carga económica. El tratamiento es sintomático y no existe un tratamiento
estándar al momento. Quizás en el futuro un tratamiento orientado hacia la etiología pudiera significar una
mejoría de las manifestaciones del espectro clínico.
Nuestro paciente reúne las características clínicas señaladas de este síndrome, como hemos podido constatar en la
literatura publicada al respecto. Solo nos queda preguntarnos ¿cuántos casos de síndrome de Angelman se
encuentran en nuestros centros de salud, en las consultas de neurología y pediatría y en las clínicas de
epilepsia que corresponden a esta enfermedad?