Como citar
Morales González MC, Peralta Rosa JN. Hipofosfatemia Ligada al X: Reporte de Caso. ADOPA. 2024;2(2):69-82. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/26
ADOPA, Vol. 2, No. 2, mayo-agosto, 2024 • ISSN (en línea): 2960-7582 • Sitio web: https://adopa.pediatriadominicana.org/index.php/adopa
Morales González MC, Peralta Rosa JN. Hipofosfatemia Ligada al X: Reporte de Caso. ADOPA. 2024;2(2):69-82. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/26
Introducción: la hipofosfatemia ligada al X se caracteriza principalmente por talla baja, raquitismo, deformidades óseas a predominio en segmento inferior del cuerpo como genu varum o genu valgum y anomalías dentales. La causa son variantes patogénicas en el gen PHEX, alterando la producción de proteínas vinculadas con la reabsorción del fosfato a nivel renal. Se transmite con patrón de herencia dominante ligado al X.
Objetivo: dar a conocer a pediatras y subespecialidades, el diagnóstico de la hipofosfatemia ligada al X en República Dominicana; promoviendo su identificación y participación oportuna en el diagnóstico y tratamiento.
Material y método: presentamos una paciente femenina de 5 años, con hallazgos fenotípicos, bioquímicos y radiográficos compatibles con esta enfermedad. Confirmada con prueba genética molecular.
Conclusiones: las manifestaciones clínicas de la paciente, su edad de presentación y su evolución previo al diagnóstico corresponden con la historia natural de esta enfermedad. Enfatizamos la importancia del reconocimiento temprano y la integración del manejo multidisciplinario, incluyendo la asesoría genética y el soporte psicológico.
Introduction: X-linked hypophosphatemia is mainly characterized by short stature, rickets, bone deformities predominantly in the lower segment of the body such as genu varum or genu valgum, and dental anomalies. The cause is pathogenic variants in the PHEX gene, altering the production of proteins linked to phosphate reabsorption at the kidney level, transmitted with an X-linked dominant inheritance pattern.
Objective: Make pediatricians and subspecialties aware of the diagnosis of x-linked hypophosphatemia in the Dominican Republic, so that they can identify it and participate in a timely manner in its diagnosis and treatment.
Material and method: We present a 5-year-old female patient with compatible phenotypic, biochemical and radiographic findings of this disease. Confirmed with molecular genetic testing.
Conclusions: The patient’s clinical manifestations, her age at presentation and her evolution prior to diagnosis correspond with the natural history of this disease. We emphasize the importance of early recognition and the integration of multidisciplinary management, including genetic counseling and psychological support.
La hipofosfatemia ligada al cromosoma X (HLX) o raquitismo hipofosfatémico ligado al cromosoma X (RHLX) es una enfermedad genética rara (OMIM: #307800) causada por disminución de la reabsorción tubular renal del fosfato; de expresión clínica muy variable y multisistémica, con una prevalencia mundial estimada en 1 por cada 20 000 nacidos vivos1-3.
Descrita por primera vez en 1937 por Albright, Butler y Bloomberg. Representa la forma más frecuente de hipofosfatemia hereditaria (80 %), caracterizada por raquitismo, osteomalacia, talla baja, deformidades óseas y anomalías dentarias4, 5.
Ocurre por diversas variantes (>460) que generan inactivación del gen PHEX (en inglés: phosphate regulating endopeptidase analog, X-linked), en el locus Xp22.1; que sintetizan endopeptidasas, enzimas que participan en la degradación de las hormonas peptídicas que se expresan predominantemente a nivel óseo y dental6-8.
El mecanismo exacto etiopatogénico de la HLX no es bien conocido aún. La pérdida de función del gen PHEX produce un aumento del factor de crecimiento de fibroblastos 23 (FGF-23), el cual a su vez reduce la expresión de los co-transportadores de sodiofosfato tipo II (NaPi-2a y NaPi-2c), principal responsable de la reabsorción del fosfato en el túbulo proximal renal (70 %), conduciendo a su disminución e hiperfosfaturia. Además de afectar la transcripción de los genes CYP27B1 y CYP24A1, que sintetizan las enzimas 1 alfa-hidroxilasa y 24 hidroxilasa respectivamente; lo que disminuye la 1,25 dihidroxivitamina D circulante (metabolito activo de la vitamina D) y la absorción intestinal de fosfato y calcio, alterando la mineralización ósea normal y siendo responsable de las manifestaciones clínicas típicas de la enfermedad9.
A continuación, presentamos un caso clínico diagnosticado por criterios clínicosradiográficos y confirmado con test molecular dirigido de una paciente asistida en la Unidad de Genética del Hospital Infantil Dr. Robert Reid Cabral, centro de referencia nacional en atención pediátrica en la República Dominicana.
Se trata de paciente femenina de 5 años, dominicana, quien a partir de los 18 meses inicia con deformidades óseas a predominio en miembros inferiores y talla baja para su grupo de edad. Con antecedentes personales patológicos de infecciones respiratorias agudas, amigdalitis recurrentes e hipertrofia de adenoides.
Hija de padres no consanguíneos, dominicanos, G2 C2, con chequeos prenatales regulares, antecedentes de infección vaginal en 1er trimestre, tratada y remitida. Teratógenos negados y sonografías obstétricas sin hallazgos patológicos. Producto único, obtenido vía cesárea, con llanto espontáneo, peso 7 6/16 libras, a término, Apgar 8/9; ameritando 7 días de ingreso por síndrome de distrés respiratorio.
Desarrollo psicomotor adecuado para edad. Asiste a educación regular. Al examen físico: talla baja desproporcionada, asimetría corporal, macrocefalia, dolicocefalia, prominencia frontal, hipertelorismo, hipoplasia facial media, puente nasal bajo, nariz prominente, filtrum corto y pronunciado, anomalías dentarias, barbilla triangular, pabellones auriculares grandes, cuello corto, asimetría torácica, ligero tórax excavatum, RsCsRs, no soplos audibles, pulmones normoventilados, no estertores agregados, abdomen semigloboso, no visceromegalias, anomalías esqueléticas múltiples en articulación hombros, cadera, rodillas, extremidades superiores e inferiores, hipermovilidad articular, dismetría en miembros inferiores, genu valgum, superposición dedos en los pies, algunas máculas hipercrómicas residuales generalizadas en piel.
Entre los estudios de laboratorios: Hemograma con GB 8,2, Hb 136.1, Hto 39.1 %, Plaq 452. Calcio: 10.1 mg/dL. Fósforo: 2.24 mg/dL, Magnesio: 1.28 mg/ Dl, Microalbúmina: Negativo, Creatinina: 0.44 mg/Dl, Urea: 21.8 mg/Dl, 25 Hidroxivitamina D : 31.8 mg/Dl, PTH: 45.6 mg/dL.
Las pruebas complementarias de imágenes reportaron: TAC cráneo, Radiografía de Tórax PA, sonografía abdominal y pélvica sin hallazgos patológicos. Ecocardiograma con insuficiencia mitral trivial.
Los hallazgos radiográficos en panorámica de miembros inferiores: tibias valgas, miembro inferior izquierdo 2.0 cm menor que contralateral, condicionado por importante genu valgum ipsilateral expresado en desviación positiva de eje de carga e importante coxa vara de caderas. En la radiografía de columna lumbosacra en proyección AP y Lateral, se evidencia pérdida de alineación posterior de L4,L5 y S1. Rectificación de lordosis lumbar fisiológica y cifosis dorsal, con cierre incompleto espinoso de L5 con relación a espina bífida oculta. En radiografía de caderas AP y rana se muestran los fémures de aspecto cortos y ensanchados de manera generalizada.
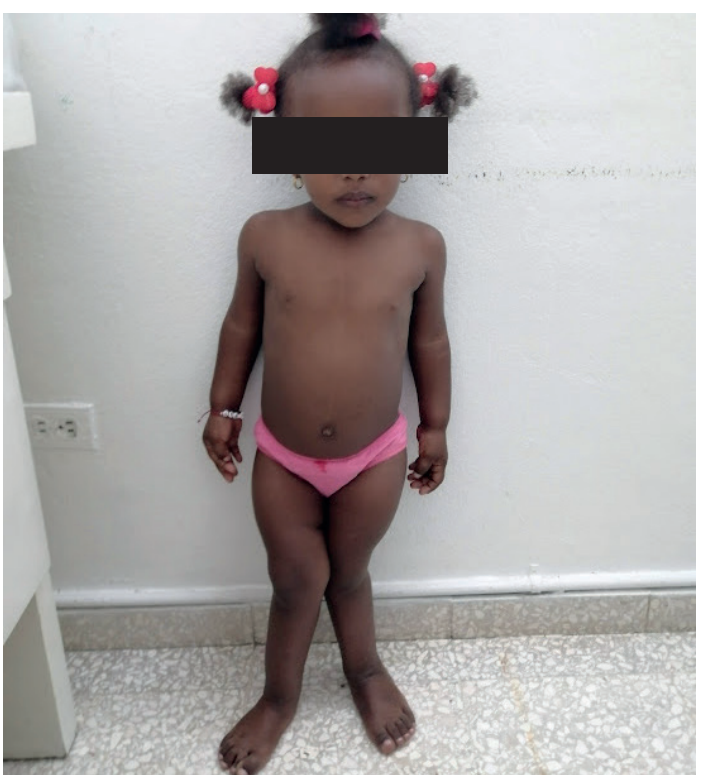
Figura 1. Vista de frente
Fuente: Expediente clínico. Hospital Infantil Dr. Robert Reid Cabral.

Figura 2. Vista dorsal
Fuente: Expediente clínico. Hospital Infantil Dr. Robert Reid Cabral.
Se evidencia baja talla, asimetría corporal, acortamiento de extremidades, anomalías óseas múltiples, deformidad en genu valgum a predominio derecho de miembros inferiores.
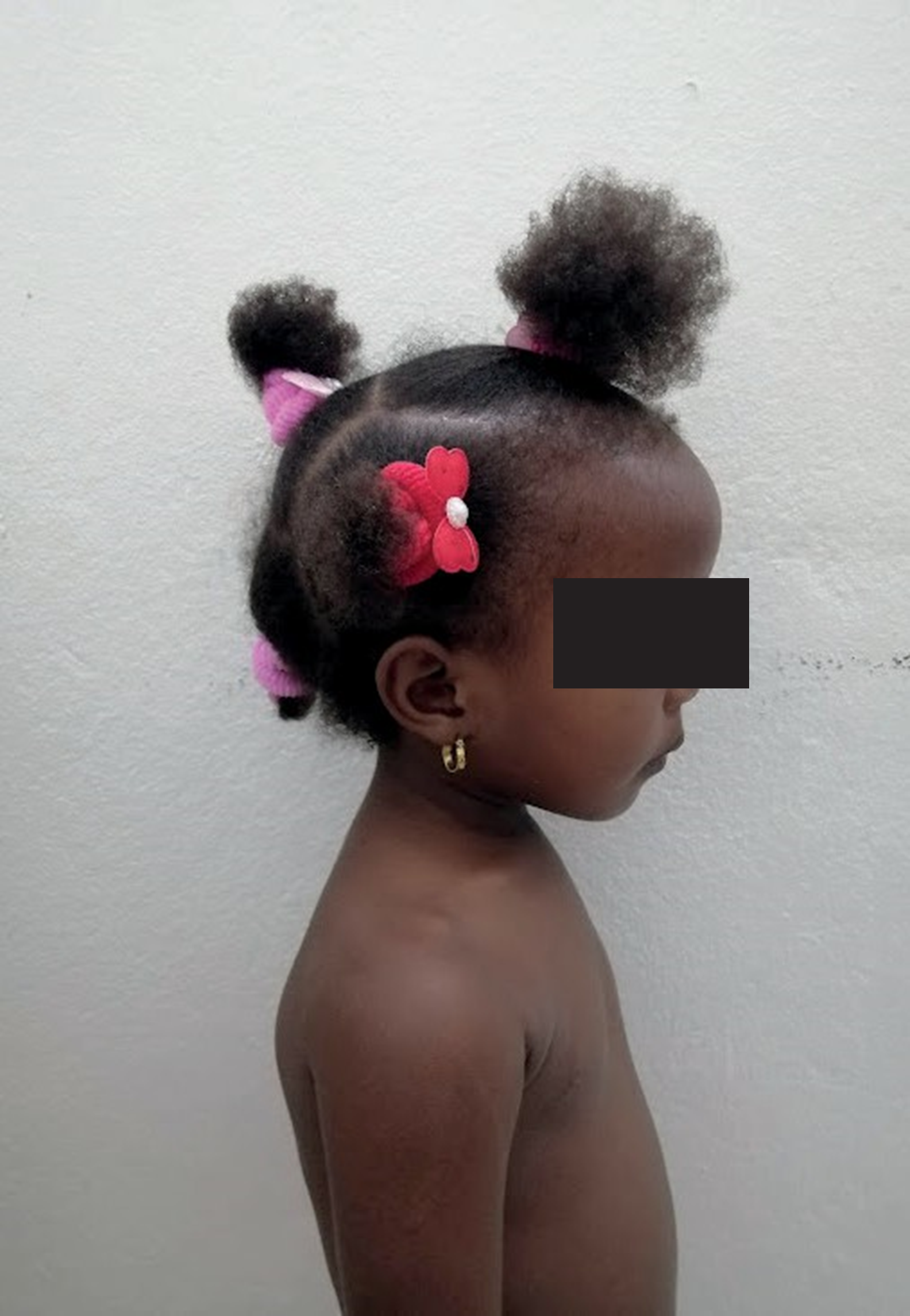
Figura 3. Vista de perfil
Fuente: Expediente clínico. Hospital Infantil Dr. Robert Reid Cabral.
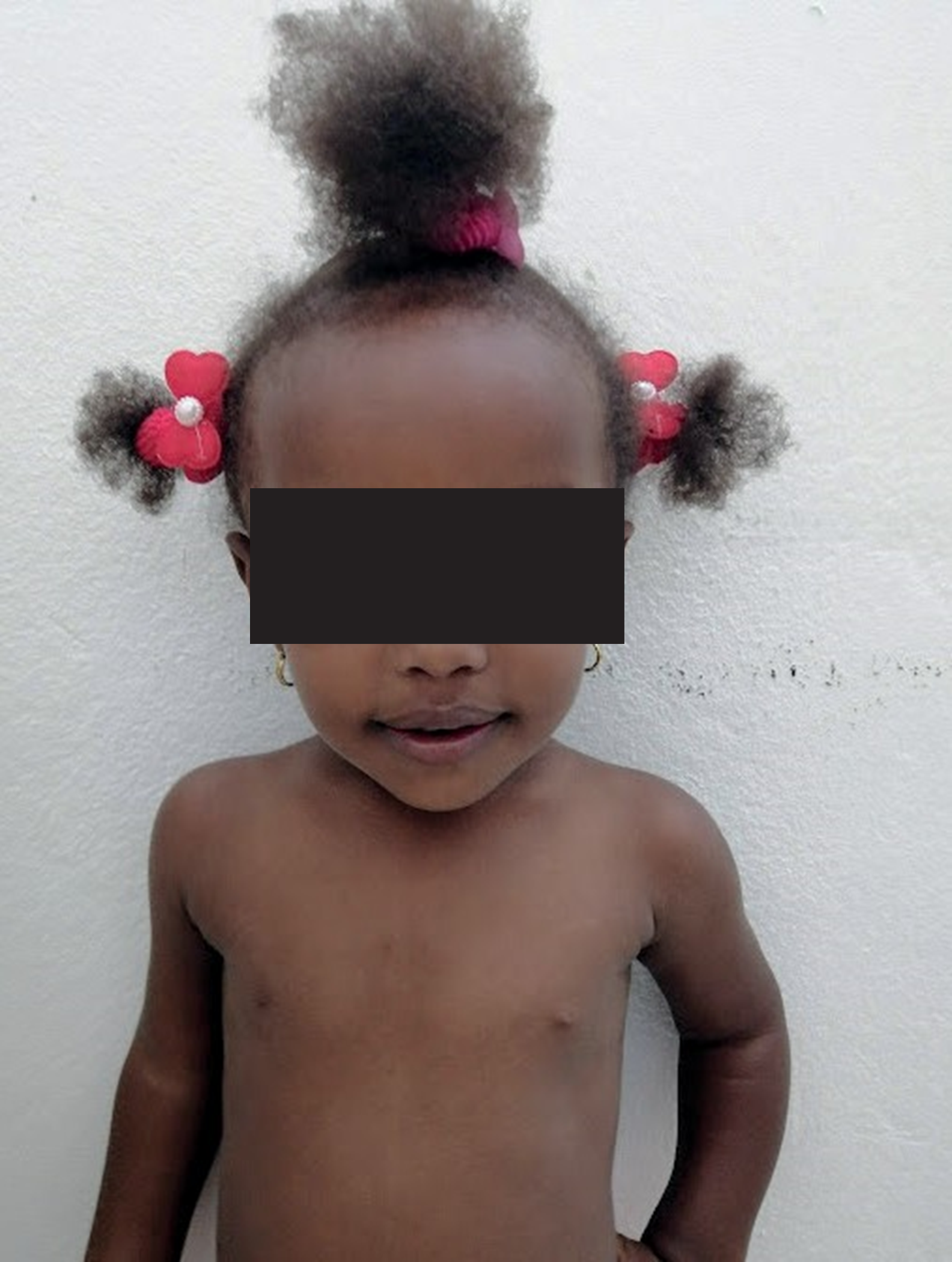
Figura 4: Vista frontal
Fuente: Expediente clínico. Hospital Infantil Dr. Robert Reid Cabral.
Se evidencian anomalías craneofaciales: macrocefalia, dolicocefalia, prominencia frontal, barbilla triangular.
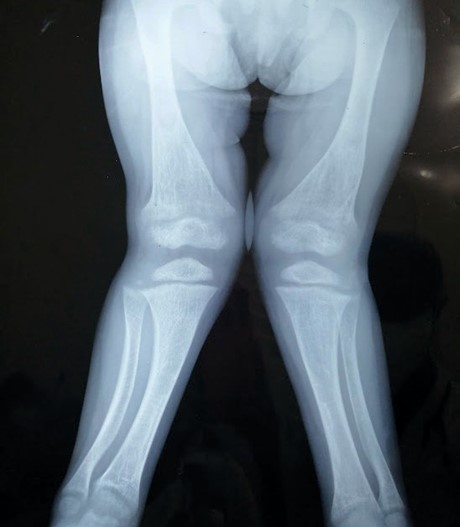
Figura 5. Rx Ambos Miembros Inferiores AP
Fuente: Expediente clínico. Hospital Infantil Dr. Robert Reid Cabral.
Deformidad en valgo con presencia de alteración de la morfología de las metáfisis femorales, tibiales y en ambos peronés en forma de copa, con disminución de la densidad ósea principalmente en 1/3 distal de ambos fémures en relación con desmineralización de estos. Disminución de la amplitud de la línea de crecimiento a nivel femoral y tibial con visualización de los núcleos de osificación femorales, tibiales y peroneos disminuidos de tamaño.
Se confirma sospecha diagnóstica de hipofosfatemia ligada al X o raquitismo hipofosfatémico ligado al X, con prueba molecular dirigida (panel de genes) que reportó: variante patogénica identificada en Gen PHEX (c.670C>T (p. Gln224) en heterocigosis (Herencia Dominante Ligada al X). Descartándose otras patologías de diagnóstico diferencial como el raquitismo hipofosfatémico autosómico dominante y recesivo, la displasia ósea fibrosa, el síndrome de Fanconi renal, el raquitismo hipofosfatémico hereditario con hipercalciuria, la deficiencia de vitamina D y la osteomalacia inducida por tumor.
La paciente se encuentra recibiendo manejo multidisciplinario integral con ortopedia pediátrica, nefrología pediátrica, endocrinología pediátrica, nutrición, odontología, pediatría, genética, soporte psicológico familiar, entre otros. Recibe actualmente suplementos de fósforo y vitamina D vía oral, con adecuada respuesta.
La hipofosfatemia ligada al X es una enfermedad monogénica poco frecuente en la literatura médica universal10, que a pesar de tener hallazgos clínicos, bioquímicos y radiográficos muy característicos, en muchos casos, sobre todo en aquellos con menor grado de expresión (espectro fenotípico), no se diagnóstica, o se realiza de forma tardía, siendo el pronóstico y calidad de vida de los pacientes dependientes de ese manejo oportuno, integral y multidisciplinario, sobre todo en el contexto socioeconómico y sanitario de países en vías de desarrollo como la República Dominicana. Motivos por lo que se publica este caso, asistido en el Hospital Infantil Dr. Robert Reid Cabral, con el objetivo de dar a conocer el mismo y su abordaje diagnóstico-terapéutico desde el ámbito pediátrico.
Se transmite con patrón de herencia dominante ligado al cromosoma X (HDLX), correspondiendo en un 1/3 de los casos a mutaciones de novo (sin antecedentes familiares y progenitores sanos). El genotipo entre las mujeres afectadas es el de heterocigoto y en los varones hemicigoto dominante. No existe salto generacional ni transmisión directa del gen de varón a varón y un riesgo de recurrencia familiar de 50 % en cada embarazo para ambos sexos. En este sentido, y por tratarse de variantes genéticas con penetrancia completa, nuestra paciente fue considerada una mutación espontánea de novo, ya que no se reportaron antecedentes en la familia; se les otorgó asesoría genética correspondiente.
Las manifestaciones clínicas de la HLX por lo general no se evidencian al nacimiento, sino entre el 1er y 2do año de vida11-12, lo que coincide con el debut clínico de nuestra paciente a partir de los 18 meses de edad.
A nivel craneal, suelen presentar craneosinostosis, macrocefalia, dolicocefalia, prominencia frontal, aplanamiento de la base del cráneo, mayor predisposición a malformaciones tipo Chiari I (44 %), siringomielia, hipoacusia neurosensorial o de conducción (16-76 %), anomalías dentarias que aumentan riesgo de desarrollar abscesos y periodontitis (61-78 %). Además, talla baja desproporcionada por reducción en la velocidad de crecimiento y acortamiento del segmento inferior condicionado por las deformidades óseas presentes: genu varum (arqueamiento de la tibia y fémur) o menos frecuente genu valgum (angulación de la rodilla hacia la línea media), coxa vara o tibias en sable9,13-15. Encontrando en nuestra paciente anomalías esqueléticas típicas de la condición como la macrocefalia, dolicocefalia, talla baja y acortamiento de miembros, aunque difiriendo de la mayoría de los estudios publicados, que reportan el genu varum como principal deformidad en miembros inferiores y no el genu valgum, presentado por nuestra paciente.
Las pruebas bioquímicas suelen presentar como principales hallazgos: hipofosfatemia (niveles bajos de fosfato en sangre), hiperfosfaturia (aumento de la excreción de fosfato en orina), calcio en orina bajo, niveles séricos de calcio normal en sangre, 25 OH Vitamina D normal, 1,25 OH Vitamina D baja o normal, PTH normal o alta, FGF-23 y fosfatasa alcalina altas16. Siendo algunos de estos reportes de laboratorios los que brindaron soporte principalmente a nuestra sospecha diagnóstica.
En adultos predominan los dolores óseos, talla baja, artrosis, predisposición a fracturas, osteoartritis, calcificación de tendones y ligamentos, rigidez, osteomalacia, entre otros. Datos que por la edad de nuestra paciente no podemos asociar, aunque en la misma ya se evidencia dolor importante a predominio en miembros inferiores17.
El tratamiento convencional, desde hace décadas, ha consistido en la administración oral de sales de fosfato y calcitriol; que, aunque mejora algunas anomalías esqueléticas y la talla, la respuesta a la terapia depende de la gravedad del raquitismo, el apego del paciente al tratamiento (diversas dosis diarias) y la habilidad del médico tratante (reajustes de dosis y monitoreo); puede llevar a complicaciones como nefrocalcinosis e hiperparatiroidismo. Desde el 2018 se aprobó el Burosumab como tratamiento para la HLX, un anticuerpo monoclonal IgG1 completamente humano contra FGF23, que proporciona mejor calidad de vida, disminuyendo riesgo de complicaciones y discapacidad18,19. Nuestra paciente ha presentado mejoría de las deformidades de miembros inferiores tras el tratamiento clínico y dos cirugías ortopédicas correctivas, y se encuentra en proceso para fines de iniciar terapia con Burosumab a 0.8 mg/kg peso corporal vía subcutánea cada 2 semanas.
La hipofosfatemia ligada al X es una enfermedad genética, con hallazgos similares a otras patologías que producen hipofosfatemia en edad pediátrica, pero con etiología y manejo diferentes, por lo que su reconocimiento y diagnóstico oportunos permiten intervenciones dirigidas e individualizadas que mejoran calidad de vida a corto y largo plazo.
Se siguieron las normas éticas protocolarias para la publicación de casos clínicos patológicos, preservando la autonomía de nuestra paciente a través de la confidencialidad de datos y consentimiento informado.
La presente investigación no ha recibido financiación alguna.
Los autores declaran que en la publicación de este artículo no hay conflicto de interés entre las partes.