Introducción
El síndrome CAPOS/CAOS fue descrito por primera vez en 1996 por Nicolaides y colaboradores1 como un trastorno neurológico con un patrón clínico caracterizado por ataxia cerebelosa, arreflexia, pes cavus, atrofia óptica y pérdida auditiva neurosensorial. En 2014, Demos y van Karnebeek asociaron este cuadro a una mutación recurrente en el gen ATP1A32, 3, el cual codifica la subunidad α3 de la ATPasa Na+/K+, presente principalmente en neuronas del cerebelo, nervio óptico, cóclea y vías motoras.
A pesar de compartir gen mutado, CAPOS/CAOS forma parte de un espectro más amplio de encefalopatías relacionadas al gen ATP1A3, como la hemiplejía alternante, la ataxia de inicio rápido y la distonía-parkinsonismo de inicio súbito4, 5. Debido a su rareza, su diagnóstico suele ser tardío o erróneamente atribuido a enfermedades como encefalitis o síndromes neuromusculares adquiridos6.
Objetivo
Presentar un caso clínico con diagnóstico genético confirmado de síndrome CAPOS/CAOS y realizar una revisión de la literatura actual, destacando las manifestaciones clínicas tempranas y la importancia del diagnóstico diferencial en encefalopatías infantiles complejas.
Presentación de caso
Se trata de un paciente masculino de 3 meses de edad, sin antecedentes personales conocidos hasta el momento. Hijo de madre de 31 años, quien durante la gestación presentó neumonía por COVID-19 a las 11 semanas, siendo hospitalizada y requiriendo cerclaje por amenaza de parto prematuro. Producto único de la tercera gestación, nacido por cesárea a las 37 semanas con llanto al nacer, permaneció 10 días en la unidad neonatal por neumonía connatal. A los pocos días de vida presentó episodios caracterizados por movimientos tónicos en el miembro superior derecho, desviación ocular, cianosis perioral y ausencia de respuesta a estímulos durante aproximadamente 10 minutos. Fue manejado inicialmente en emergencia con diazepam (0.2 mg/kg/d) y oxígeno suplementario, e ingresado para control de crisis. Posteriormente, presentó varios episodios de neumonía y un ingreso a la unidad de cuidados intensivos por estatus epiléptico.
Al examen físico destacó hipotonía generalizada con posición en “libro abierto”, ausencia de sostén cefálico, reflejo plantar de Babinski positivo, clonus inducido, reflejo de Galant y paracaídas positivos, presión palmar y plantar 2/4, y reflejos osteotendinosos 3/4. Dentro de los estudios complementarios se evidenció una TSH en 8.1 mIU/L, por lo que se inició tratamiento con levotiroxina 25 mcg, logrando descenso a 0.03 mIU/L, lo que motivó su suspensión. Además, se detectó serología positiva para Epstein-Barr IgG y se identificó una estenosis parcial del esófago medio (véase Figura 1).
La resonancia magnética cerebral mostró hipoplasia del cuerpo calloso, mega cisterna magna y una variante del síndrome de Dandy-Walker (Figuras 2A y 2B). Los estudios neurofisiológicos (electroencefalograma, electromiograma y pruebas auditivas) no arrojaron hallazgos significativos. Ante la persistencia de hallazgos clínicos, se solicitó un estudio genético de miopatías mitocondriales (ADN mitocondrial más panel de genes nucleares mitocondriales), el cual resultó negativo. Posteriormente, se amplió la evaluación genética con un panel de atrofias ópticas sindrómicas, donde se identificó una mutación en heterocigosis en el exón 18 del gen ATP1A3 (NM_152296) c.2452G>A; p.Glu818Lys, descrita como patogénica en bases de datos como CLINVAR y responsable del síndrome CAPOS/CAOS, con herencia autosómica dominante2. El estudio de segregación familiar confirmó que se trataba de una mutación de novo, correspondiendo así a un caso esporádico.
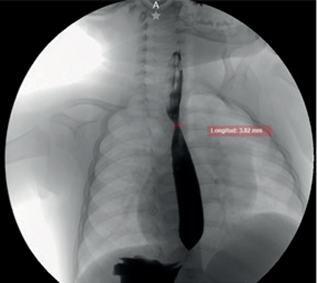
Figura 1. Estudio esofagograma en proyección anteroposterior que muestra una estenosis parcial del esófago medio, evidenciada por una zona de estrechamiento luminal segmentario con paso filiforme del contraste
Fuente: expediente clínico del paciente.
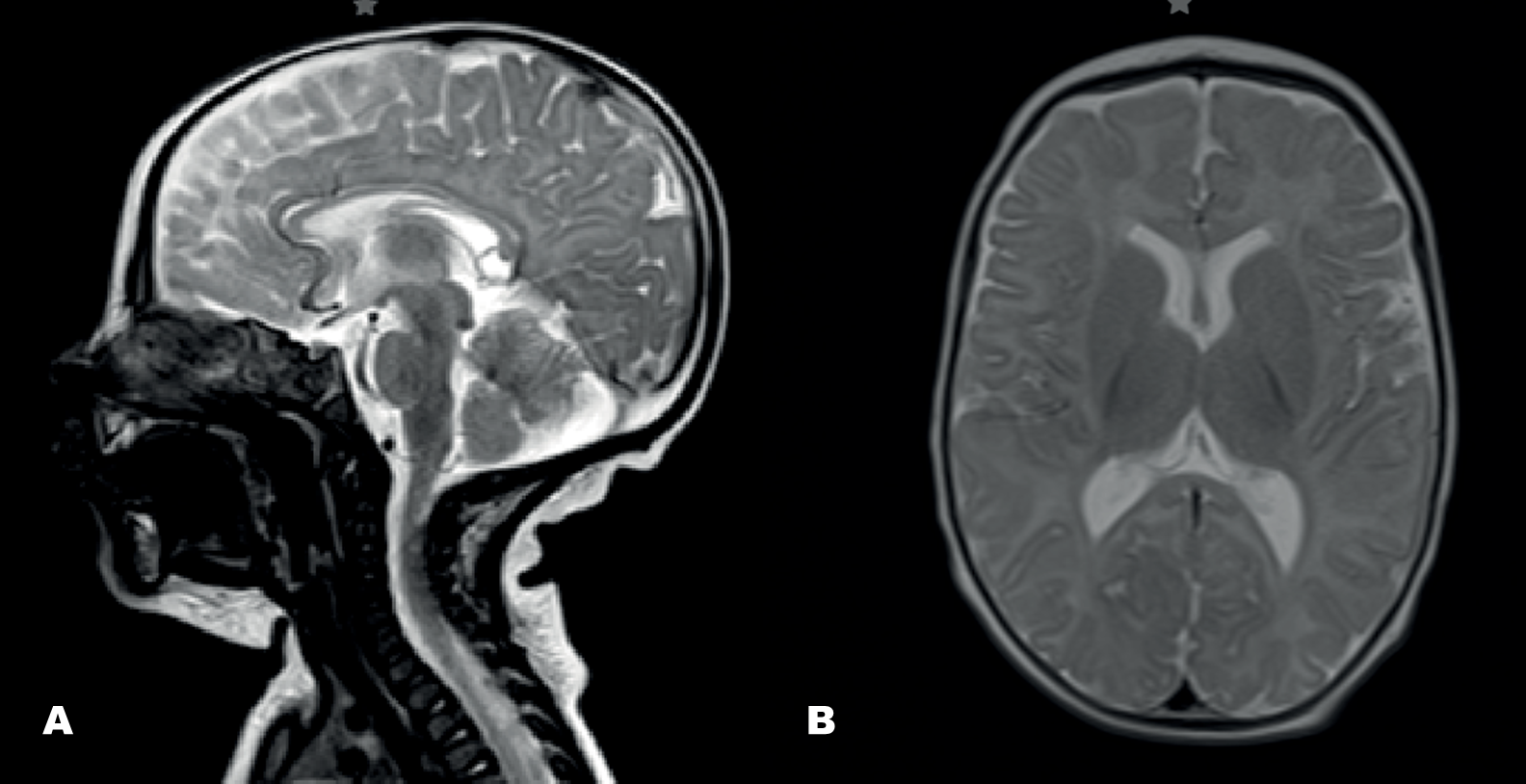
Figura 2. Imágenes por resonancia magnética cerebral en cortes sagital (Figura A) y axial (Figura B), secuencia T2
Fuente: expediente clínico del paciente.
Se observa hipoplasia del cuerpo calloso, presencia de una mega cisterna magna y hallazgos compatibles con una variante del síndrome de Dandy-Walker, caracterizada por vermis cerebeloso hipoplásico y agrandamiento de la fosa posterior.
Discusión
El síndrome CAPOS/CAOS (OMIM#601338) es una entidad poco frecuente, descrita inicialmente por Nicolaides et al. en 1996, cuyo acrónimo refleja las principales manifestaciones clínicas: cerebellar ataxia, areflexia, pes cavus, optic atrophy y sensorineural hearing loss1. En 2014, Demos y Van Karnebeek asociaron este síndrome con la variante heterocigota c.2452G>A (p.Glu818Lys) en el gen ATP1A32.
El gen ATP1A3 codifica la subunidad α3 de la ATPasa Na+/K+, una enzima esencial para el mantenimiento del gradiente iónico transmembrana, con expresión predominante en la cóclea, nervio óptico, corteza cerebelosa y fibras nerviosas que inervan el músculo esquelético. Esta distribución tisular explica muchas de las manifestaciones clínicas del síndrome3.
Además del síndrome CAPOS/CAOS, que representa una de las manifestaciones fenotípicas menos frecuentes, se han descrito múltiples síndromes neurológicos vinculados a mutaciones en ATP1A3, tales como la hemiplejia alternante de la infancia (AHC; OMIM#614820) y la distonía-parkinsonismo de aparición rápida (DYT12; OMIM#128235)4. En los últimos años se han reportado nuevos fenotipos relacionados, incluyendo encefalopatía epiléptica infantil temprana, encefalopatía recurrente con ataxia cerebelosa (RECA), ataxia de aparición rápida, crisis autonómicas precoces y distonía asimétrica paroxística. Este espectro en expansión ha llevado a algunos autores a proponer que se trata de un continuum fenotípico, más que de entidades alélicas distintas5.
Usualmente, los síntomas comienzan en la infancia y se desencadenan tras episodios febriles. Durante estas crisis, los pacientes presentan ataxia cerebelosa, encefalopatía y debilidad muscular. Con frecuencia, estas manifestaciones agudas se confunden con encefalitis o variantes atípicas del síndrome de Guillain-Barré6, 7. Aunque la recuperación es común tras los episodios febriles, algunos pacientes presentan secuelas neurológicas parciales. Los casos reportados refieren entre uno y tres episodios agudos durante la evolución clínica.
Un hallazgo clínico variable es la presencia de pies cavos, ausente hasta en el 70 % de los casos, como se observa en nuestro paciente. Esta variabilidad ha llevado a algunos expertos a preferir el término CAPOS/CAOS en lugar de CAPOS, para incluir aquellos casos sin esta característica ortopédica8. Asimismo, es frecuente que las pruebas complementarias no revelen alteraciones miopáticas ni neuropáticas, situación que también se confirmó en este caso.
Dada la heterogeneidad fenotípica del síndrome y el solapamiento clínico con otras entidades, es fundamental realizar un adecuado diagnóstico diferencial con atrofias ópticas sindrómicas dominantes y trastornos de la cadena respiratoria mitocondrial. Actualmente, no existe un tratamiento específico para el síndrome CAPOS/CAOS9. Se ha sugerido el uso de acetazolamida durante los episodios febriles agudos, con el objetivo de reducir el déficit neurológico asociado. El posible beneficio de este fármaco se basa en su capacidad para inhibir la anhidrasa carbónica, lo cual genera una disminución del pH extracelular, favoreciendo el funcionamiento de la ATPasa Na+/K+9, 10.
Conclusión
El síndrome CAPOS/CAOS, aunque raro, representa una condición clínica relevante asociada con mutaciones en el gen ATP1A3. La identificación temprana de sus manifestaciones, que incluyen ataxia inducida por fiebre, encefalopatía y debilidad muscular, es crucial para el diagnóstico adecuado y el manejo de los pacientes. A pesar de que las pruebas neurofisiológicas complementarias pueden no revelar hallazgos significativos, el análisis genético sigue siendo fundamental para confirmar la mutación patogénica, como se observó en nuestro caso. La mutación ATP1A3 en el exón 18 es un hallazgo clave para establecer el diagnóstico definitivo, en este caso, con una herencia autosómica dominante. Aunque no existe un tratamiento específico para el síndrome, los enfoques terapéuticos como el uso de acetazolamida pueden ser prometedores en la prevención de los episodios agudos. Dada la diversidad de fenotipos asociados a las mutaciones de ATP1A3, es fundamental que los profesionales de la salud mantengan un alto índice de sospecha y realicen un diagnóstico diferencial con otras condiciones neurológicas y mitocondriales.
Financiamiento
Los autores declaran que no recibieron fondos específicos de agencias de financiación en los sectores público, comercial o sin ánimo de lucro para la realización de esta investigación.
Conflictos de interés
Lo autores declaran no tener ningún conflicto de interés.
Referencias
- Nicolaides P, Appleton RE, Fryer A. Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS): a new syndrome. Journal of Medical Genetics [Internet]. 1996;33(5):419–21. Available from: https://pubmed.ncbi.nlm.nih.gov/8733056/
- Maas RPPWM, Schieving JH, Schouten M, Erik-Jan Kamsteeg, P.C B. The Genetic Homogeneity of CAPOS Syndrome: Four New Patients With the c.2452G>A (p.Glu818Lys) Mutation in the ATP1A3 Gene. Pediatric Neurology [Internet]. 2016;59:71-75.e1. Available from: https://pubmed.ncbi.nlm.nih.gov/27091223/
- Demos MK, van Karnebeek CD, Ross CJ, Adam S, Shen Y, Zhan SH, et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet Journal of Rare Diseases [Internet]. 2014;9(1):15. Available from: https://pubmed.ncbi.nlm.nih.gov/24468074/
- Duat Rodríguez A, Prochazkova M, Santos Santos S, Rubio Cabezas O, Cantarin Extremera V, González-Gutiérrez-Solana L. Early Diagnosis of CAPOS Syndrome Before Acute-Onset Ataxia—Review of the Literature and a New Family. Pediatric Neurology [Internet]. 2017 Jun;71:60–4. Available from: https://pubmed.ncbi.nlm.nih.gov/28483396/
- Sabouraud P, Riquet A, Spitz MA, Deiva K, Nevsimalova S, Mignot C, et al. Relapsing encephalopathy with cerebellar ataxia are caused by variants involving p.Arg756 in ATP1A3. European Journal of Paediatric Neurology [Internet]. 2019;23(3):448–55. Available from: https://pubmed.ncbi.nlm.nih.gov/30862413/
- Heimer G, Sadaka Y, Israelian L, Feiglin A, Ruggieri A, Marshall CR, et al. CAOS—Episodic Cerebellar Ataxia, Areflexia, Optic Atrophy, and Sensorineural Hearing Loss. Journal of Child Neurology [Internet]. 2015;30(13):1749–56. Available from: https://pubmed.ncbi.nlm.nih.gov/25895915/
- Shang P, Zhu M, Wang Y, Zheng X, Wu X, Zhu J, et al. Axonal variants of Guillain–Barré syndrome: an update. Journal of Neurology. 2020;268(7):2402–19. Available from: https://pubmed.ncbi.nlm.nih.gov/32140865/
- Salles PA, Mata IF, Brünger T, Lal D, Fernandez HH. ATP1A3-Related Disorders: An Ever-Expanding Clinical Spectrum. Frontiers in Neurology [Internet]. 2021;12. Available from: https://pubmed.ncbi.nlm.nih.gov/33868146/
- Kostopoulou E, Avgeri A, Apostolou MI, Tzifas S, Dimitriou G. A novel presentation of an ATP1A3 gene mutation case report and literature review. European Review for Medical and Pharmacological Sciences [Internet]. 2022;26(4). Available from: https://pubmed.ncbi.nlm.nih.gov/35253165/
- Sankhyan N, Sharawat I, Kasinathan A, Suthar R. CAPOS syndrome: A rare ATP1A3-related disorder. Annals of Indian Academy of Neurology [Internet]. 2020;23(3):397. Available from: https://journals.lww.com/annalsofian/fulltext/2020/23030/capos_syndrome_a_rare_atp1a3_related_disorder.51.aspx