Introducción
La dermatomiositis juvenil (DMJ) es una enfermedad autoinmune rara, caracterizada principalmente por una inflamación crónica de los músculos estriados y lesiones cutáneas características. Se considera la miopatía inflamatoria idiopática más común en la población pediátrica y constituye un desafío diagnóstico y terapéutico significativo en la práctica clínica pediátrica1. La DMJ se manifiesta habitualmente en niños alrededor de los siete años de vida, con una incidencia aproximada de dos a cuatro casos por millón de niños al año, siendo más frecuente en el sexo femenino2.
La etiología de la dermatomiositis juvenil es multifactorial. Se ha propuesto que la combinación de predisposición genética y factores ambientales, como infecciones virales, desencadena una respuesta inmune anómala que resulta en daño vascular e inflamación muscular3. En cuanto a sus manifestaciones clínicas, la DMJ se presenta típicamente con debilidad muscular proximal simétrica de inicio insidioso, junto con lesiones cutáneas patognomónicas como el exantema heliotropo y las pápulas de Gottron2. También pueden observarse telangiectasias periungueales, calcinosis cutánea, úlceras y, en algunos casos, compromiso sistémico que incluye afecciones gastrointestinales, cardíacas o pulmonares2, 4.
Diversas opciones de tratamiento están disponibles para la DMJ, siendo el metotrexato y los esteroides el tratamiento estándar para los pacientes recién diagnosticados5. Sin embargo, la mayoría de los fármacos no han sido evaluados en ensayos clínicos debido a la rareza de la enfermedad5. Dentro de las opciones terapéuticas en casos severos se encuentra la inmunoglobulina humana intravenosa (IGIV), en especial en aquellos pacientes con manifestaciones dermatológicas prominentes5.
En el presente reporte de caso se describe una paciente pediátrica con dermatomiositis juvenil, destacando sus manifestaciones clínicas, hallazgos diagnósticos y evolución terapéutica, con el objetivo de resaltar la importancia del diagnóstico y tratamiento para evitar complicaciones y asegurar una mejor calidad de vida en estos pacientes.
Este reporte de caso se elaboró siguiendo las directrices CARE.
Descripción del caso
Se trata de paciente femenina nacida en el 2017, quien a los tres años experimentó su primer episodio de caída sobre su propio plano de sustentación mientras realizaba actividades cotidianas, según refiere la madre. Sin embargo, no es hasta inicios del 2023, cuando la paciente presenta un segundo episodio de caída aunado a dolor y debilidad generalizada acompañado de pérdida del conocimiento, que decide llevarla al centro de salud de su comunidad. En dicho centro la manejan por el trauma, descartan lesiones cerebrales y al observar mejoría, refieren a consulta de pediatría. A pesar del manejo y de las evaluaciones por consultas, los síntomas persistieron, y la madre refiere aparición progresiva de lesiones dermatológicas eritematosas y violáceas que se generalizaron al resto del cuerpo y edema palpebral. Pese al tratamiento dermatológico no especificado, la madre no observó mejoría, el cuadro de debilidad general empeoró, agregándose artralgias, disnea, tos y rigidez articular generalizada en un período menor a seis meses. Por lo cual decide acudir a consulta de reumatología.
Al examen físico, se encontró una paciente disneica, con hallazgos significativos de eritema heliotropo, eritema en región cervical sugestivo de signo de la Chal, pápulas y placas descamativas en las articulaciones metacarpofalángicas e interfalángicas de ambas manos que dan la impresión de pápulas de Gottron (véase Figura 1), lesiones hipopigmentadas en extremidades superiores e inferiores y tronco, además de sarcopenia generalizada.
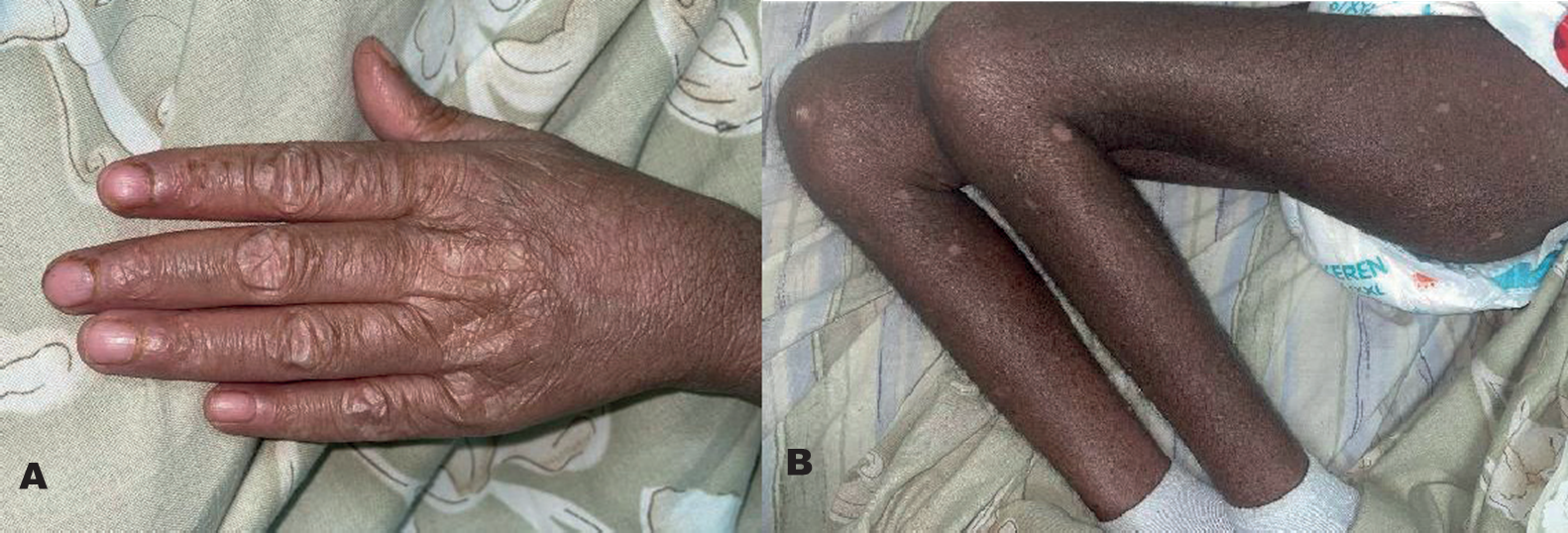
Figura 1. A: mano izquierda que evidencia lesiones elevadas, eritematosas o hipopigmentadas confirmatorias de pápulas de Gottrón, con aspecto atrófico en las articulaciones metacarpofalángicas e interfalángicas. B: extremidades inferiores con marcada sarcopenia
Fuente: archivo personal de los autores. Foto de la paciente.
Luego de la evaluación inicial en la consulta, se decidió su ingreso para realizar exámenes y analíticas con fines diagnósticos. Durante su ingreso se realizó una biopsia muscular donde se encontró infiltrado linfocítico perivascular en vasos del perimisio del tejido muscular, atrofia y necrosis de fibras musculares. La electromiografía fue concluyente para potenciales de unidades motoras polifásicas de corta duración y pequeña amplitud con irritabilidad insercional. Ambos estudios son concluyentes para dermatomiositis inflamatoria.
Se realizaron analíticas de lugar, encontrando aumento de aspartato aminotransferasa (AST) 51 U/L, lactato deshidrogenasa (LDH) 1147 U/L y creatina quinasa (CPK) 657 UI. Debido a estos hallazgos clínicos, analíticos, biopsia y electromiografía, se confirma el diagnóstico de dermatomiositis juvenil por criterios de EULAR/ACL. Además, la disnea se acompañó de hallazgos como roncos y crepitantes pulmonares. La radiografía mostró un patrón intersticial bilateral, confirmando el diagnóstico de neumonía.
La paciente, de 5 años, 18 kg y 0.79 m² de superficie corporal, ingresó recibiendo ácido fólico 0.3 mg/kg/dosis (en jarabe, 5 mL/24 h), hidroxicloroquina 200 mg VO cada 24 h, metotrexato subcutáneo 15 mg/m² (18.9 mg) semanal y una dosis 15 g de terapia con inmunoglobulina humana IV (IgGIV), administrada inicialmente a 5 g en 48 h y posteriormente 10 g en 24 h. Una neumonía bilateral requirió posponer la inmunoglobulina por una semana, tiempo durante el cual se instauró tratamiento con cefazolina 100 mg/kg/día (600 mg IV c/8 h), ceftriaxona 1 g (675 mg IV c/12 h), hidrocortisona 30 mg IV c/12 h y acetaminofén 180 mg VO c/6 h. Tras la resolución del cuadro respiratorio, la paciente mostró buena tolerancia y evolución favorable con el tratamiento ambulatorio.
En febrero de 2025, sin embargo, la paciente se reingresó por debilidad muscular, empeoramiento de artralgia y de las lesiones dermatológicas que le impedía realizar sus actividades diarias. Durante esta hospitalización se reinició el tratamiento con inmunoglobulina IV a una dosis de 35 g y se sustituyó el metotrexato por micofenolato de mofetilo 3 g VO c/24 h. Al alta, se estableció un esquema de mantenimiento que incluía L-carnitina en jarabe 5 mL c/24 h, hidroxicloroquina a misma dosis, suplemento combinado de zinc (7.5 mg), calcio (300 mg) y vitamina D (100 UI) en suspensión de 5 mL tras cada comida, cetirizina en jarabe 10 mg/10 mL (5 mL cada noche), micofenolato de mofetilo 500 mg VO c/12 h, prednisolona 10 mg VO c/12 h y aplicación tópica de crema con urea tres veces al día. Dado que la familia no contaba con los recursos para mantener la terapia ambulatoria, se retomó el esquema con metotrexato. Semanas después, la paciente acudió nuevamente por empeoramiento clínico, lo que motivó una nueva hospitalización.
Actualmente, la paciente acudió de nuevo a consulta de reumatología por empeoramiento de su debilidad general y dolor articular, por lo que fue hospitalizada para recibir inmunoglobulina IV a la misma dosis previa y manejo sintomático de su dolor y fatiga. Al ingreso se encontraba afebril, con signos vitales dentro de parámetros estables. Presentaba: FR 28 rpm, FC 100 lpm, TA 90/60 mmHg, talla 116 cm (P15), peso 16.82 kg (P3), SatO2 98 %.
Al examen físico se mostraban lesiones dermatológicas de base, aunado a hallazgos de poiquilodermia (véase Figura 2), rigidez articular inferior y superior en mayor intensidad en las articulaciones interfalángicas, metacarpofalángicas edematizadas y dolorosas, sarcopenia marcada, cambios inflamatorios periungueales, a la auscultación roncus y murmullo vesicular disminuido. Con estos hallazgos, se procedió a su ingreso para el control de la patología de base y la optimización de su tratamiento inmunomodulador.
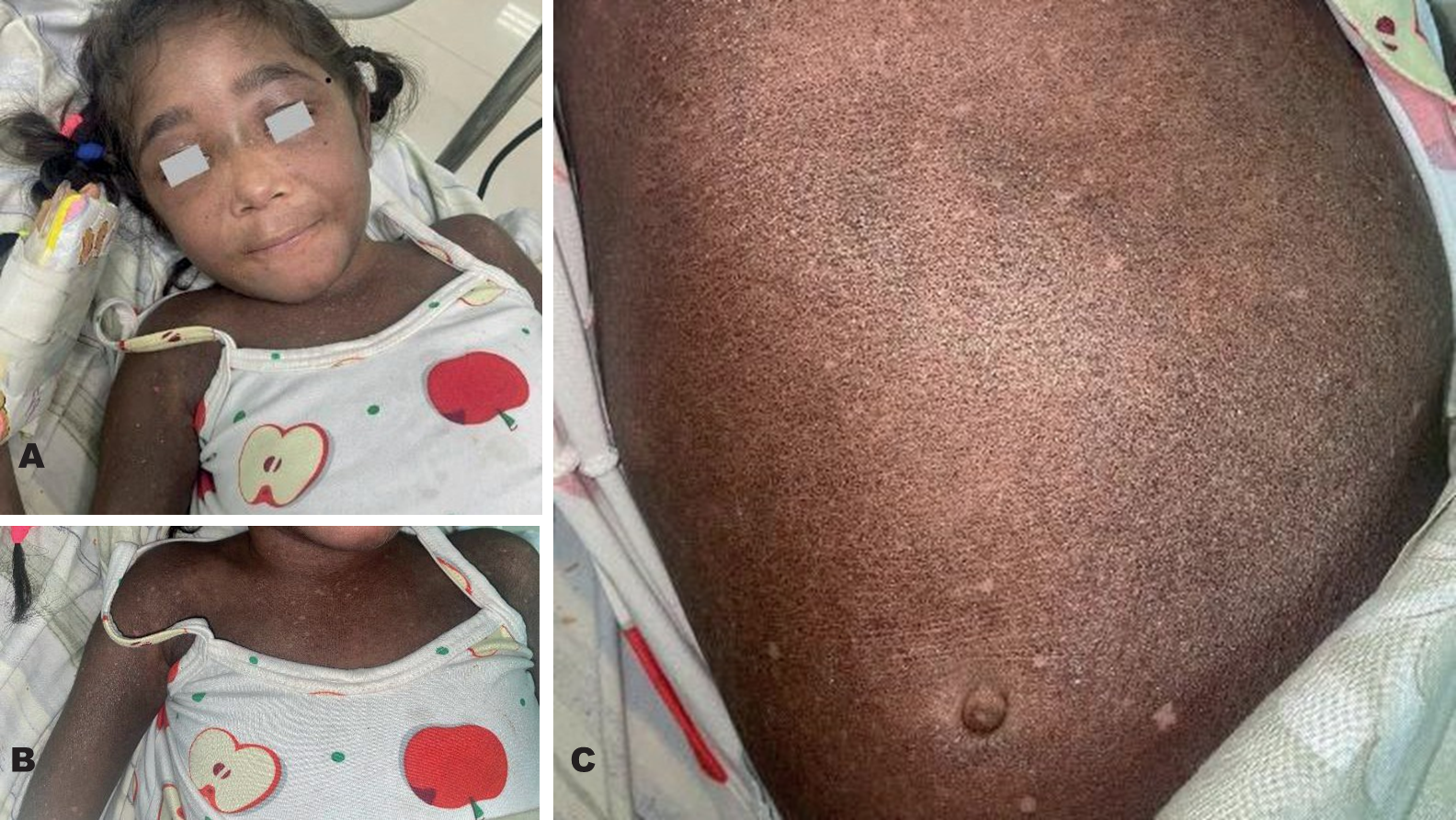
Figura 2. A: hiperpigmentación generalizada acompañada de máculas hipopigmentadas más datos de atrofia cutánea y telangiectasias sugestivas de poiquilodermia, que constituyen un patrón de daño crónico en la piel. Además, se evidencia el eritema heliotropo en los párpados de la paciente. B: abdomen con mejor vista de los datos de poiquilodermia. C: signo de Chal
Fuente: archivo personal de los autores. Foto de la paciente.
Antecedentes patológicos de la niñez
Antecedentes no patológicos
- Alergias: negadas.
- Antecedentes quirúrgicos: negados.
- Traumatismos: negados.
- Transfusiones negados.
- Esquema de vacunación: incompleto según refiere la madre, no muestra tarjeta.
- Hábitos tóxicos: negados.
Antecedentes familiares
- Madre viva, aparentemente sana.
- Padre vivo, aparentemente sano.
- Abuelos y abuelas vivos, materna con HTA, paterno con DM2, además aparentemente sanos.
- Prima por parte de familia materna con diagnóstico de lupus eritematoso sistémico.
Evaluación diagnóstica
La evaluación diagnóstica inicial se basó en el examen físico, la medición de las medias antropométricas y la historia clínica obtenida. Se realizaron analíticas de lugar entre ellas hemograma, tipificación, glucemia, LDH, CPK, transaminasas, examen de orina, azoados y una radiografía de tórax.
Tabla 1. Biometría hemática y química sanguínea
| Hemograma |
| RBC | 4.68e6/µL - 4.0 – 5.50e6/µL |
| HCT | 39.9 % - 37.0 – 50.0 % |
| HGB | 12.70 g/dL - 11.0 – 16.0 g/dL |
| MCV | 85.4 fL - 82 – 100 fL |
| MCH | 27.2 pg - 27.0 – 32.0 pg |
| MCHC | 32.90 g/dL - 32.0 – 37.0 g/dL |
| WBC | 6.92e3/µL - 4.5 – 11.0e3/µL |
| NEUT%* | 39.9% - 40 – 75% |
| PLAQ | 469e3/µL - 150 – 450e3/µL |
| LYMPH% | 40.90 % - 37 – 72.0 % |
| Química sanguínea |
| Glicemia | 84.4 mg/dL - 70 – 105 mg/dl |
| Creatinina | 0.4 mg/dL - 0.7 – 1.4 mg/dl |
| Urea | 12.9 mg/dL - 15 – 45 mg/dL |
| Bun | 6.0 mg/dL - 6 – 21 mg/dL |
| AST | 37 U/L - 0 – 31 U/L |
| ALT | 13.2 U/L - 0 – 32 U/L |
| Albumina | 4.2 mg/dL - 3.5 – 5.0 mg/dL |
| LDH | 1,147 U/L - 230 – 460 U/L |
| Electrolitos |
| Calcio | 8.80 mg/dL - 10.0 – 12.0 mg/dL |
| Cloro | 106 mmol/L - 98 – 107 mmol/L |
| Fósforo | 5.2 mg/dL - 4.0 – 7.0 mg/dL |
| Magnesio | 1.6 mg/dL - 1.6 – 2.60 mg/dL |
| Potasio | 4.88 mmol/L - 3.5 – 5.3 mmol/L |
| Sodio | 139.1 mmol/L - 135 – 148 mmol/L |
Nota. Valores normales; valor mínimo – valor máximo
Fuente: expediente clínico.
Los análisis muestran un hemograma (véase Tabla 1) dentro de parámetros normales, salvo una ligera trombocitosis. La función renal se encuentra conservada, aunque con creatinina y urea ligeramente bajas, probablemente relacionadas con baja masa muscular o dieta. Las enzimas hepáticas indican una elevación leve de AST y un aumento marcado de LDH, lo que sugiere daño celular, de origen muscular. La glucemia y los electrolitos están mayormente normales, excepto por un leve descenso del calcio sérico, sin datos de hipocalcemia clínicamente significativa (véase Tabla 1). Estos hallazgos ameritan seguimiento clínico, en especial para investigar la actividad de la patología de base.
Asimismo, los hallazgos en el examen físico como taquipnea, palidez conjuntival, manifestaciones dermatológicas variadas como eritema, hiperpigmentación e hipopigmentación y lesiones atróficas que respetan la comisura nasolabial. Murmullo vesicular disminuido, rigidez y dolor en extremidades inferiores, así como edema articular y lesiones nodulares en las interfalángicas, hablan de la cronicidad del trastorno autoinmune que presenta la paciente. Además, en la radiografía de tórax (véase Figura 3) existen hallazgos de infiltrado con patrón intersticial difuso en ambos campos pulmonares, los cuales, en conjunto con la clínica, son sugestivos de neumonía bilateral.
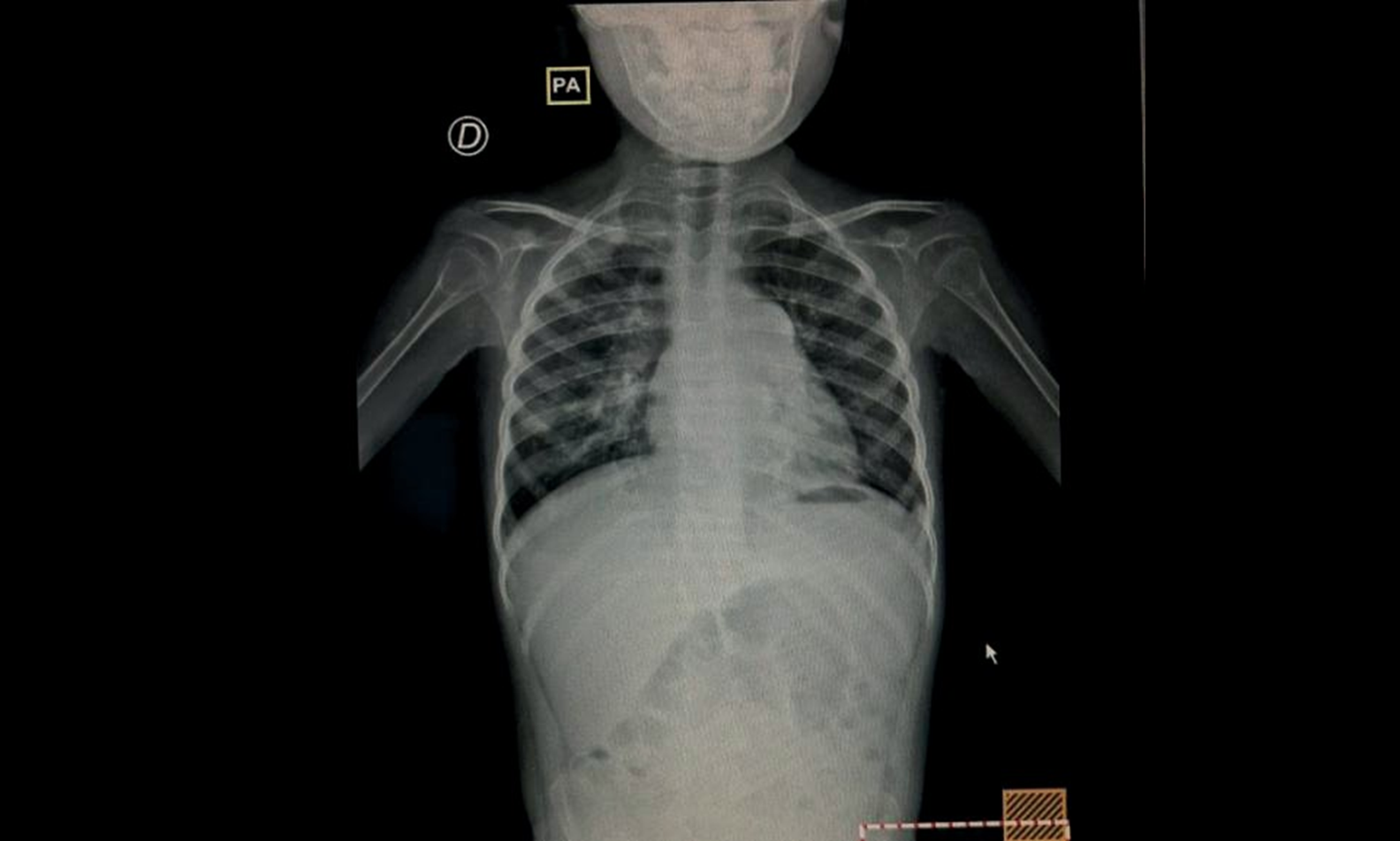
Figura 3. Radiografía de tórax posteroanterior con hallazgos de infiltrado con patrón intersticial difuso en ambos campos pulmonares
Fuente: expediente clínico.
Intervención terapéutica
A su llegada a la emergencia, la paciente fue ingresada a urgencias pediátricas para vigilancia clínica y estabilización, mientras se preparaba para la administración de la inmunoglobulina humana. Se mantuvo su medicación de base en forma ambulatoria y se añadió una solución mixta al 0.9 % 1000 mL c/24 h (45 mL c/1 h) para un Holliday Segar al 80 % y ketorolaco 30 mg (9 mg c/8 h) IV a dosis de 0.5 mg/kg/dosis. La inmunoglobulina se planteó dar 2 g/kg/ dosis (34 g) a pasar 1/3 de la dosis dentro de las primeras 48 horas, y los otros 2/3 a pasar en las 24 horas posteriores. Además, se realizó interconsulta con fisiatría, nutrición y cardiología. Como medidas generales se mantuvieron vigilancia bajo monitor de signos vitales, revisión del estado general, estado hemodinámico, diuresis y datos de dolor.
Seguimiento y resultados
La paciente fue ingresada en sala de urgencia pediátrica, donde se realizaron las analíticas de lugar, resultando dentro de los parámetros normales. Se realizó una radiografía de tórax, no encontrando hallazgos patológicos. Debido a su historia de disnea, cardiología recomendó la realización de un ecocardiograma y una tomografía de tórax para valoración de parénquima pulmonar. Dichas recomendaciones fueron pautadas para realizar de forma ambulatoria. Durante los próximos tres días, se administró el medicamento de alto costo sin complicaciones adicionales. La paciente fue egresada de la unidad de urgencia sin eventualidades, además de reducción de debilidad general y dolor articular.
Discusión
La dermatomiositis juvenil (DMJ) es una enfermedad autoinmune sistémica rara, caracterizada por inflamación muscular y lesiones cutáneas patognomónicas, con una incidencia de dos a cuatro casos por millón de niños al año y un predominio en el sexo femenino6. El caso aquí presentado es representativo de la forma clásica de DMJ con afectación cutánea prominente, debilidad muscular simétrica de inicio insidioso y evolución a compromiso pulmonar intersticial.
La paciente presentó múltiples manifestaciones clínicas típicas como pápulas de Gottron, eritema heliotropo, signo del chal y poiquilodermia, hallazgos consistentes con reportes recientes que destacan estas lesiones como claves diagnósticas en DMJ7. Asimismo, la debilidad muscular progresiva y los hallazgos en la electromiografía y biopsia muscular refuerzan la sospecha clínica, cumpliendo con los criterios de EULAR/ACR y Bohan y Peter8, 9. La presencia de disnea, crepitantes pulmonares y patrón intersticial en la radiografía de tórax sugiere afectación pulmonar, una complicación cada vez más reconocida en pacientes con anticuerpos específicos como anti-MDA5, incluso en ausencia inicial de síntomas respiratorios10.
Desde el punto de vista fisiopatológico, se ha identificado una vasculopatía inmunomediada como mecanismo clave, donde se produce una activación aberrante de linfocitos T y B dirigida contra el endotelio capilar de piel y músculo, lo que lleva a isquemia, inflamación y daño tisular progresivo4, 11. Esta microangiopatía inflamatoria se manifiesta clínicamente como debilidad muscular simétrica, lesiones cutáneas características (como el exantema heliotropo y pápulas de Gottron) y, en casos más severos, afectación pulmonar y gastrointestinal. En nuestra paciente, la presencia de lesiones cutáneas extensas, edema palpebral, debilidad muscular progresiva y signos de neumonía intersticial se explican por este patrón vasculopático multisistémico típico de la DMJ activa.
La dermatomiositis juvenil, aunque infrecuente, puede asociarse a complicaciones graves y potencialmente mortales, en especial a nivel respiratorio y gastrointestinal12. Se ha descrito disfonía y disfagia por debilidad faríngea, así como hipoventilación e insuficiencia respiratoria secundaria a afectación del diafragma, lo que representa una urgencia médica12. Por otro lado, la vasculopatía gastrointestinal severa puede derivar en edema, úlceras y perforaciones de la pared gastrointestinal13. A su vez, tanto pancreatitis como hepatitis se han asociado a la enfermedad, necesitando hacer un diagnóstico diferencial extenso ante la presencia de dolor abdominal14. En general, el compromiso gastrointestinal severo se ha asociado a una mayor actividad de la enfermedad y mortalidad en estos pacientes13. A pesar de que se han descrito manifestaciones gastrointestinales asociadas a la DMJ, es importante tomar en cuenta que el uso de glucocorticoides a altas dosis se asocia a diversas complicaciones endocrinológicas, musculoesqueléticas y gastrointestinales que pueden presentarse en estos pacientes15.
Además, ciertos autoanticuerpos específicos de miositis (MSA), como anti-MDA5 o anti-NXP2, se han vinculado con fenotipos clínicos particulares. Por ejemplo, los anticuerpos anti-MDA5 se asocian a afectación pulmonar precoz e intersticial y enfermedad cutánea severa sin elevación significativa de CPK16, 17. Aunque en este caso no se identificaron dichos autoanticuerpos por limitaciones diagnósticas, el compromiso pulmonar intersticial, la marcada debilidad y las lesiones ulceradas sugieren un perfil clínico que podría corresponder a un subtipo asociado a MSA de mal pronóstico, como el anti-MDA5. Este componente autoinmune específico es importante porque predice la respuesta al tratamiento y el riesgo de recaídas.
Desde el punto de vista terapéutico, el manejo inicial incluyó metotrexato e inmunoglobulina intravenosa (IgIV), abordaje respaldado por múltiples estudios que destacan la eficacia de esta combinación para inducir remisión y reducir recaídas, en comparación con monoterapia con esteroides18, 19. La recurrencia de los síntomas, especialmente cutáneos y articulares, a pesar del tratamiento, motivó la sustitución temporal del metotrexato por micofenolato de mofetilo, otro inmunosupresor utilizado en casos refractarios con buenos resultados documentados20.
Cabe destacar que la interrupción del tratamiento con micofenolato por motivos económicos y el regreso al metotrexato evidencian una realidad frecuente en países de ingresos medios, donde el acceso sostenido a terapias de alto costo constituye una limitación importante. Esta barrera puede influir directamente en la evolución clínica, aumentando el riesgo de recaídas y daño crónico, tal como se ha reportado en la literatura21.
Una de las fortalezas del presente caso es la documentación longitudinal detallada, incluyendo parámetros clínicos, bioquímicos y radiológicos, así como la respuesta a diferentes esquemas terapéuticos. También se evidencia un enfoque multidisciplinario con participación de reumatología, nutrición, fisiatría y cardiología, considerado esencial para el manejo óptimo de la DMJ22. Entre las limitaciones, se encuentra la falta de determinación de anticuerpos específicos de miositis (como anti-NXP2 o anti-MDA5), lo cual podría haber permitido una mejor estratificación del riesgo y personalización del tratamiento. Asimismo, no se describe la evaluación funcional con escalas estandarizadas (como CMAS o MMT8), lo cual limitaría la cuantificación objetiva de la debilidad muscular y la respuesta terapéutica.
Finalmente, la perspectiva de la madre resulta fundamental. Ella refiere sentimientos de incertidumbre ante los múltiples cambios terapéuticos y la dificultad económica para sostener medicamentos de alto costo como la inmunoglobulina. A pesar de esto, destaca la mejoría clínica de su hija y su deseo de continuar con el tratamiento siempre que sea posible. Esta experiencia refleja la importancia del acompañamiento emocional, la educación continua y la toma de decisiones compartida en enfermedades crónicas pediátricas.
Conclusión
Este caso de dermatomiositis juvenil ilustra la importancia del diagnóstico temprano, el seguimiento multidisciplinario y la adaptación individualizada del tratamiento según la respuesta clínica y el contexto socioeconómico del paciente. Las manifestaciones clásicas cutáneo-musculares, junto con el compromiso pulmonar, reflejan la naturaleza multisistémica de la enfermedad y la necesidad de vigilancia continua. El uso de inmunoglobulina intravenosa y metotrexato, seguido de ajustes con micofenolato de mofetilo en fases de recaída, evidencia un enfoque terapéutico dinámico que ha permitido controlar la actividad de la enfermedad y mejorar la calidad de vida de la paciente. No obstante, las limitaciones de acceso a fármacos de alto costo resaltan el desafío adicional que enfrentan muchos pacientes en contextos vulnerables. Este caso subraya la relevancia de considerar factores médicos, psicosociales y económicos en la toma de decisiones clínicas, promoviendo una atención pediátrica integral y centrada en la familia.
Referencias
- Leung A, Lam J, Alobaida S, Leong K, Wong A. Juvenile Dermatomyositis: Advances in Pathogenesis, Assessment, and Management. Current pediatric reviews [Internet].
- Gara S, Jamil RT, Muse ME, Litaiem N. Juvenile Dermatomyositis. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 [citado 20 de abril de 2025]. Disponible en:
https://www.ncbi.nlm.nih.gov/books/NBK534236/
- Arakkal A, Peter P, Cherian V, Joy R. Juvenile dermatomyositis in a 6-year-old girl: A case report. Indian Journal of Case Reports [Internet].
- Papadopoulou C, McCann LJ. The Vasculopathy of Juvenile Dermatomyositis. Front Pediatr. 9 de octubre de 2018;6:284.
- Doudouliaki T, Papadopoulou C, Deakin CT. Use of Rescue Therapy with IVIG or Cyclophosphamide in Juvenile Myositis. Curr Rheumatol Rep. abril de 2021;23(4):24.
- Juvenile dermatomyositis: advances in clinical presentation, myositis-specific antibodies and treatment | World Journal of Pediatrics [Internet]. [citado 21 de mayo de 2025]. Disponible en:
https://link.springer.com/article/10.1007/s12519-019-00313-8
- Owen ED, Choy EH, Piguet V. Juvenile dermatomyositis: new clinical trial evidence to underpin therapeutic shared decision making. British Journal of Dermatology. 1 de octubre de 2016;175(4):665-6.
- Okka K, Belghazi M, Dehimi A, Benarab Z, Bouabdallah S, Bioud B. P052 Juvenile dermatomyositis, a multi-faceted disease. Rheumatology. 1 de noviembre de 2021;60(Supplement_5):keab722.044.
- Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. EULAR/ACR Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and their Major Subgroups. Ann Rheum Dis. diciembre de 2017;76(12):1955-64.
- He L. Recent research on myositis-specific autoantibodies in juvenile dermatomyositis. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics. 2021;23 10:1064-8.
- Wienke J, Deakin CT, Wedderburn LR, van Wijk F, van Royen-Kerkhof A. Systemic and Tissue Inflammation in Juvenile Dermatomyositis: From Pathogenesis to the Quest for Monitoring Tools. Front Immunol [Internet]. 18 de diciembre de 2018 [citado 21 de mayo de 2025];9. Disponible en:
https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.02951/full
- Rhim JW. Juvenile Dermatomyositis. J Rheum Dis. 1 de enero de 2022;29 (1):14-21.
- Xiangyuan C, Xiaoling Z, Guangchao S, Huasong Z, Dexin L. Juvenile dermatomyositis complications: navigating gastrointestinal perforations and treatment challenges, a case report. Front Pediatr [Internet]. 12 de julio de 2024 [citado 30 de mayo de 2025];12. Disponible en:
https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2024.1419355/full
- Besnard C, Gitiaux C, Girard M, Galmiche-Rolland L, Talbotec C, Quartier P, et al. Severe Abdominal Manifestations in Juvenile Dermatomyositis. J Pediatr Gastroenterol Nutr. febrero de 2020;70(2):247-51.
- Koshi EJ, Young K, Mostales JC, Vo KB, Burgess LP. Complications of Corticosteroid Therapy: A Comprehensive Literature Review. J Pharm Technol. diciembre de 2022;38(6):360-7.
- Honda M, Shimizu F, Sato R, Nakamori M. Contribution of Complement, Microangiopathy and Inflammation in Idiopathic Inflammatory Myopathies. Journal of Neuromuscular Diseases. 2 de enero de 2024;11(1):5-16.
- Kobayashi I. Advances in Juvenile Dermatomyositis: Pathophysiology, Diagnosis, Treatment and Interstitial Lung Diseases—A Narrative Review. Children. Septiembre de 2024;11(9):1046.
- Muramatsu K, Ujiie H, Yokozeki M, Tsukinaga I, Ito M, Shikano T, et al. Recurrence of juvenile dermatomyositis 8 years after remission. JAAD Case Reports. 1 de enero de 2017;3(1):29-32.
- Ayad N, Gherbi M, Gacem O, Falek H, Labboun L, Amirat L, et al. 82 Juvenile dermatomyositis with early-onset anti-MDA 5 antibodies: a case report. Rheumatology. 1 de octubre de 2022;61(Supplement_2):keac496.078.
- Çakan M, Karadağ ŞG, Ayaz NA. Complete and sustained resolution of calcinosis universalis in a juvenile dermatomyositis case with mycophenolate mofetil. The Turkish Journal of Pediatrics. 25 de octubre de 2019;61(5):771-5.
- Polat MC, Altaş ,Meryem Hilal, Öden Akman ,Alkım, Tehçi ,Ali Kansu, Ardıçlı ,Didem, Çelikel Acar ,Banu, et al. Rare skin manifestation of juvenile dermatomyositis: peri-orbital oedema and facial swelling. Paediatrics and International Child Health. 1 de octubre de 2024;44(3-4):141-5.
- Sulaiman W, Mohd Lepatoni F, Tang JJ, Baharudin NB. Juvenile clinically amyopathic dermatomyositis (CADM): Case-based review. The Egyptian Rheumatologist. 1 de junio de 2023;45(3):203-7.