Introduction
Galactosemia is an inborn error of metabolism with autosomal recessive inheritance that disrupts the Leloir pathway, which converts galactose to glucose-1-phosphate. Classic galactosemia (Type I), the most severe presentation, is caused by biallelic pathogenic variants in the GALT gene (chromosome 9p13.3)1, 2. The resulting loss of enzymatic activity leads to the accumulation of galactose-1-phosphate, galactitol, and galactonate, which cause cellular toxicity affecting the liver, kidneys, lens, and central nervous system.
In settings without early detection programs, the disease presents as a potentially fatal neonatal metabolic emergency2, 4. A significant challenge is that 25% to 30% of patients do not carry the common mutations included in standard targeted molecular panels5-8. This report describes the case of a Dominican female infant whose diagnosis was confirmed through comprehensive molecular analysis following a negative initial mutation panel.
Case description
A 2-month and 10-day-old female infant was referred for evaluation due to progressive hepatic involvement and abdominal distension. Symptom onset occurred in the first week of life with persistent jaundice and severe indirect hyperbilirubinemia (22.41 mg/dL), requiring hospitalization and a blood transfusion.
Post-discharge, the infant developed progressive abdominal distension, lower extremity edema, and spider nevi (telangiectasias). She was initially treated with antivirals for presumed congenital cytomegalovirus (CMV) and Epstein-Barr virus (EBV) infection but showed no clinical improvement.
Physical Examination Admission
General: Hemodynamically stable but exhibiting failure to thrive (ponderal and statural undergrowth).
Skin: Pale-grayish skin tone with mildly icteric sclerae.
Abdomen: Distended; liver palpable 10 cm below the right costal margin with spider nevi on the abdominal wall.
Extremities: Bilateral lower extremity edema.
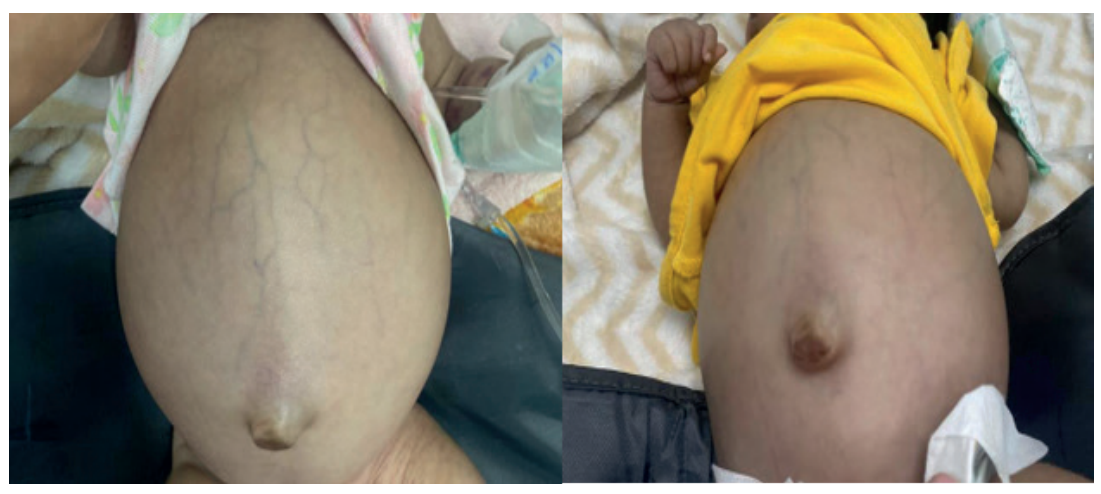
Figure 1. Clinical photographs of the patient demonstrating abdominal distension with spider angiomas upon arrival at the center
Source: medical record.
Diagnostic evaluation
The diagnostic workup was conducted in a stepwise manner, integrating newborn screening, molecular confirmation, biochemical studies, and clinical correlation, in accordance with the recommendations of the International Clinical Guideline for the Management of Classical Galactosemia3.
Biochemical Screening
Expanded newborn screening at 2 months and 10 days of age via capillary blood sampling on dried blood spot filter paper (PerkinElmer – StepOne Program), showed a GALT activity of 17 µM (Reference: >40 µM). Notably, total galactose levels were within normal limits (4.6 mg/dL), illustrating that enzymatic activity is a more sensitive screening parameter than total serum galactose1, 3, 5.
Molecular Analysis
Etiological confirmation was achieved via buccal cell sequencing (Prevention Genetics, PGxome method), identifying two heterozygous variants in the GALT gene:
- c.425T>A (p.Met142Lys): Classified as pathogenic.
- c.292G>C (p.Asp98His): Classified as likely pathogenic.
The co-occurrence of these variants is consistent with compound heterozygosity for classic galactosemia; however, allelic phase (cis/trans configuration) could not be determined in the absence of parental studies. Additionally, two variants of uncertain significance (VUS) were identified in the GALM gene (c.956G>A and c.391C>T), whose clinical relevance remains undetermined in the current context3, 5, 6.
Laboratory findings
Table 1. Complete Blood Count (Admission)
| Parameter |
Result |
Interpretation |
| Leukocytes |
16.110 /µL |
Elevated |
| Neutrophils |
36.7% |
Diminished |
| Lymphocytes |
54.5% |
Elevated |
| Hemoglobin (Hb) |
8.1 g/dL |
Anemia |
| Hematocrit (Hct) |
29.7% |
Diminished |
| MCV |
117.3 fL — macrocytosis |
Elevated |
| Platelets |
90.000 /µL |
Thrombocytopenia |
Note. Reference values for infants under 12 months.
Source: Hospital Infantil Dr. Arturo Grullón Laboratory.
Table 2. Blood Chemistry (Admission)
| Test |
Result |
Reference Range |
Interpretation |
| Potassium |
4.6 mEq/L |
3.4–4.5 |
Mildly elevated |
| Chloride |
103 mEq/L |
98–106 |
Normal |
| Total bilirubin |
6.8 mg/dL |
0.1–1.2 |
Elevated |
| Direct bilirubin |
0.3 mg/dL |
0.0–0.2 |
Elevated |
| Indirect bilirubin |
6.49 mg/dL |
≤1.0 |
Elevated |
| SGOT (AST) |
116 U/L |
0–38 |
Elevated |
| ALT (SGPT) |
43 U/L |
15–45 |
Upper limit of normal |
| Albumin |
2.7 g/dL |
3.5–5.0 |
Hypoalbuminemia |
| Urea |
38.3 mg/dL |
15–45 |
Normal |
| BUN |
17.9 mg/dL |
6–21 |
Normal |
| Creatinine |
0.2 mg/dL |
0.7–1.4 |
Low for age |
| TP / TPT |
Normal |
— |
Normal |
Note. PT: prothrombin time; PTT: partial thromboplastin time.
Source: medical record.
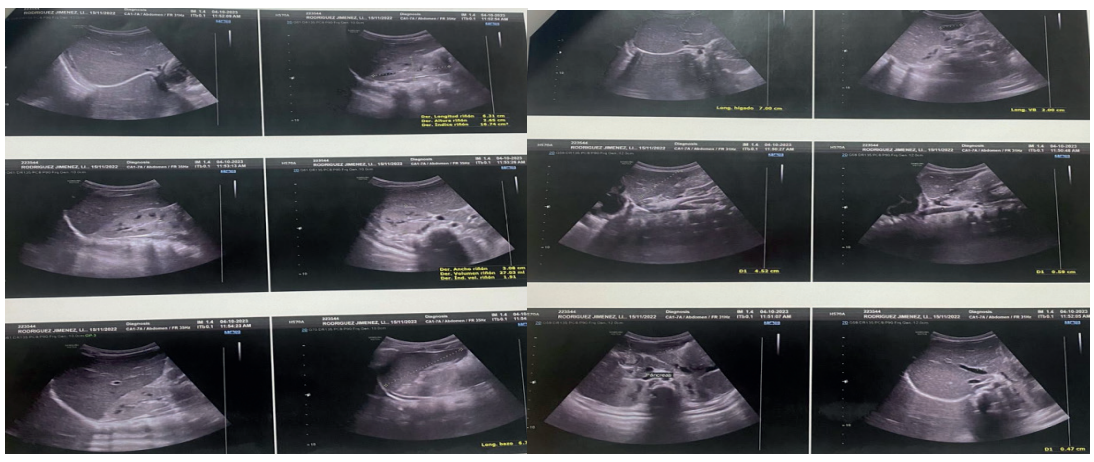
Figure 2. Abdominal sonograms with hepatic predominance
Source: medical record.
Therapeutic intervention and follow-up
Following confirmation of the type I galactosemia diagnosis, a lactose-free, soy-based infant formula was initiated. Supportive management included fluid balance maintenance, lactulose, and furosemide for edema control. Micronutrient supplementation (vitamins, zinc, and folic acid) was also provided.
Evolution
7 Months: Ultrasound showed normalization of hepatic size and resolution of ascites, though splenomegaly and mild renal changes persisted.
10 Months: Splenomegaly resolved. The patient demonstrated progressive reduction of jaundice and stabilization of her general condition.
Long-term: The patient requires ongoing longitudinal multidisciplinary surveillance to monitor late complications like cognitive decline or premature ovarian insufficiency.
Discussion
This case highlights the diagnostic challenges in settings without universal expanded newborn screening. The dissociation between GALT enzymatic activity and normal total serum galactose is a recognized source of false negatives in many screening programs1, 3, 5, 9. Furthermore, the initial negative mutation panel illustrates that more than 300 pathogenic variants exist, many of which are not included in common targeted panels5, 10.
The temporal factor is critical; international guidelines mandate that galactose restriction should begin upon suspicion, as delay prolongs exposure during a period of high organ vulnerability3, 4, 11. While dietary restriction reverses acute symptoms, the risk of long-term complications persists regardless of treatment promptness2, 4.
Conclusion
Type I galactosemia should be actively considered in infants presenting with persistent jaundice, hepatomegaly, and failure to thrive, even when total galactose levels are normal and initial mutation panels are negative. Reduced GALT enzymatic activity is the most sensitive diagnostic trigger. This case documents a favorable outcome through timely dietary restrictions and underscores the urgent need for expanded newborn screening programs.
References
- Berry GT. Classic galactosemia and clinical variant galactosemia. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2000 [updated 2023]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1518/
- Succoio M, Sacchettini R, Rossi A, Parenti G, Ruoppolo M. Galactosemia: biochemistry, molecular genetics, newborn screening, and treatment. Biomolecules. 2022;12(7):968. Disponible en: https://pubmed.ncbi.nlm.nih.gov/35883524/
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40(2):171-6. Disponible en: https://doi.org/10.1007/s10545-016-9990-5
- Therrell BL, Padilla CD. Current status of newborn bloodspot screening worldwide 2024: a comprehensive review of recent activities (2020-2023). Int J Neonatal Screen. 2024;10(2):38. Disponible en: https://doi.org/10.3390/ijns10020038
- Cantley NWP, Barski P, Kemp H, Hogg SL, Wu HYT, Bowron A, et al. Incidental detection of classical galactosemia through newborn screening: a 10-year analysis. Int J Neonatal Screen. 2023;10(1):2. Disponible en: https://doi.org/10.3390/ijns10010002
- Hong X, He M. Newborn screening for classic galactosemia: biochemical and molecular perspectives. OBM Genet. 2022;6(3):161. Disponible en: https://doi.org/10.21926/obm.genet.2203161
- Panis B, van Erven B, Rubio-Gozalbo ME. Brain function in classic galactosemia: molecular and clinical insights. Front Genet. 2024;15:1355962. Disponible en: https://doi.org/10.3389/fgene.2024.1355962
- Alaee M, Saneifard H, Shakiba M, Hanifeh M, Moarefian S. Unusual presentation of classical galactosemia: importance of early diagnosis and dietary treatment. Clin Case Rep. 2025. Disponible en: https://doi.org/10.1002/ccr3.70170
- Pereira D, Loftus E, Thompson CE, Boyle F, McNulty J, Boruah R, Crushell E, et al. Clinical and developmental outcomes after long-term newborn screening for classical galactosemia. JIMD Rep. 2025. Disponible en: https://pmc.ncbi.nlm.nih.gov/articles/PMC12105914/
- Wang Y, Li H, Zhang X, et al. Classic galactosemia with novel GALT variants: genotype-phenotype correlation. BMC Pediatr. 2024;24:469. Disponible en: https://doi.org/10.1186/s12887-024-04769-0
- Chen HA, Hsu CL, Lin WD, et al. Twelve-year review of newborn screening and incidence of galactosemia. Mol Genet Metab. 2024;142(1):45-52. Disponible en: https://doi.org/10.1016/j.ymgme.2024.02.003