Como citar
Morales González MC, Marranzini De León C. Ataxia-telangiectasia: primer reporte de caso clínico en la República Dominicana. ADOPA. 3(2):39-48. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/62
ADOPA, Vol. 3, No. 2, mayo-agosto, 2025 • ISSN (en línea): 2960-7582 • Sitio web: https://adopa.pediatriadominicana.org/index.php/adopa
Morales González MC, Marranzini De León C. Ataxia-telangiectasia: primer reporte de caso clínico en la República Dominicana. ADOPA. 3(2):39-48. Disponible en: https://adopa.pediatriadominicana.org/index.php/adopa/article/view/62
Introducción: la ataxia-telangiectasia es un trastorno hereditario poco frecuente (con una incidencia aproximada de 1 de cada 40,000 a 100,000 nacidos vivos a nivel mundial). También conocido como síndrome de Louis Bar en honor a la neuropsiquiatra belga Denise Louis Bar, quien lo describió por primera vez en 1941. Es una enfermedad neurodegenerativa y multisistémica que se presenta en la infancia con un espectro variable de manifestaciones clínicas que incluyen ataxia, corea, disartria, inmunodeficiencia, telangiectasias en ojos y piel y predisposición a ciertos tipos de cáncer. Causado por variantes patogénicas bialélicas en homocigosis o heterocigosis compuesta en el gen ATM (Ataxia-Telangiectasia Mutated) que se transmite con patrón de herencia autosómica recesiva.
Presentación del caso: presentamos el primer caso descrito en la República Dominicana de un niño de 7 años diagnosticado con ataxia-telangiectasia. El paciente presentaba signos clínicos característicos de la enfermedad como ataxia, disartria, telangiectasias oculares y cutáneas, infecciones de repetición y debilidad muscular, confirmándose con prueba genética molecular dirigida (secuenciación del gen ATM), que reportó una variante patogénica en homocigosis, ATM: c.7913G>A (p. Trp2638*).
Discusión y conclusiones: el asesoramiento genético es fundamental para el buen manejo clínico y reproductivo de los pacientes y sus familiares. La expresividad variable de esta condición conlleva una dificultad diagnóstica importante y por tanto un abordaje multidisciplinario con enfoque integral para mejorar calidad de vida y pronóstico.
Introduction: Ataxia-telangiectasia is a rare inherited disorder (with an incidence of approximately 1 in 40,000 to 100,000 live births worldwide). Also known as Louis Bar Syndrome, after the Belgian neuropsychiatrist Denise Louis Bar, who first described it in 1941, it is a neurodegenerative, multisystemic disease that presents in childhood with a variable spectrum of clinical manifestations, including ataxia, chorea, dysarthria, immunodeficiency, ocular and skin telangiectasias, and a predisposition to certain cancers. It is caused by biallelic pathogenic variants in homozygous or compound heterozygous genes in the ATM (Ataxia-Telangiectasia Mutated) gene, which are transmitted in an autosomal recessive inheritance pattern.
Case presentation: we present the first case described in the Dominican Republic of a 7-year-old boy diagnosed with Ataxia-Telangiectasia. The patient presented characteristic clinical signs of the disease, such as ataxia, dysarthria, ocular and cutaneous telangiecta- sias, recurrent infections, and muscle weakness. The diagnosis was confirmed with targeted molecular genetic testing (sequencing of the ATM gene), which reported a homozygous pathogenic variant, ATM: c.7913G>A (p.Trp2638*).
Discussion and conclusions: genetic counseling is essential for the proper clinical and reproductive management of patients and their families. The variable expressivity of this condition entails significant diagnostic difficulty and therefore requires a multidisciplinary, comprehensive approach to improve quality of life and prognosis.
La ataxia-telangiectasia (AT) o síndrome de Louis Bar, es un trastorno genético raro (OMIN #208900), Orphanet Orpha Number: ORPHA100, que produce disminución o ausencia de la proteína ATM, una quinasa que interviene en la reparación del ADN en las células del cerebelo, la conjuntiva y la piel y su respuesta a factores externos tóxicos como el daño oxidativo, los agentes alquilantes y la radiación ionizante1.
La causa de esta enfermedad son variantes patogénicas (>600), con pérdida de función en el gen de la ataxia-telangiectasia mutada ( ATM) ubicado en el cromosoma 11q22-23. Se transmite con patrón de herencia autosómica recesiva, por lo que los pacientes afectados deben recibir un alelo mutado de ambos progenitores, siendo estos portadores sanos (heterocigotos) y con un riesgo de recurrencia familiar de 25 % de hijos afectados en cada embarazo2. La presentación clínica abarca un espectro de condiciones neurodegenerativas y sistémicas, complejas y de aparición en la primera infancia (forma típica o clásica) o en menor frecuencia en etapas más tardía o en la adultez (forma atípica o leve). Las manifestaciones iniciales suelen aparecer antes de los 5 años con dificultad progresiva para la marcha y alteración del equilibrio (ataxia cerebelar), trastorno del lenguaje (disartria), sialorrea y alteración para los movimientos oculares voluntarios (apraxia oculomotora). Además de una neuropatía motora y sensitiva progresiva con pérdida de los reflejos tendinosos y alteraciones de movimiento de diferentes tipos (corea, atetosis, mioclonías, distonías). Por lo general, la inteligencia está conservada, pero se han descrito casos con trastornos de aprendizajes o discapacidad intelectual leve3.
El segundo signo que suele presentarse es la aparición de telangiectasias oculocutáneas (capilares dilatados), generalmente entre los 3-6 años; de predominio en conjuntiva bulbar, puente nasal, pabellones auriculares, cuello y en los pliegues flexores de los antebrazos.
En la mayoría de los pacientes la inmunodeficiencia primaria suele manifestarse por linfopenia grave (inmunodeficiencia combinada o hipogammaglobulinemia), con susceptibilidad de infecciones recurrentes principalmente en vías respiratorias altas y bajas: rinitis, sinusitis, otitis, bronquitis y neumonías4.
En adición, este defecto reparador del DNA tiene como consecuencia que los pacientes con AT tengan un riesgo incrementado de malignidad de alrededor de un 25 % con respecto a la población general. Los más comunes son las neoplasias hematológicas, linfomas y leucemias en edad infantil. En la adultez, además, hay riesgo de tumores sólidos como cáncer de mama, ovárico, hepático, gástrico, entre otros5.
A pesar de que los individuos heterocigotos para una variante en el gen ATM no desarrollarán AT, su riesgo de malignidad está incrementado, especialmente en las mujeres portadoras, las cuales se ha demostrado que tienen aproximadamente un riesgo 2.3 veces mayor de contraer cáncer de mama en comparación con la población general6. En consecuencia, el estudio en cascada de los progenitores y demás familiares de primer y segundo grado permite establecer un correcto manejo de cara a la familia. Para esto, es inminente proveer al paciente de un adecuado y oportuno asesoramiento genético para establecer el riesgo de enfermedad.
Dada la amplia variedad fenotípica que acompaña a este trastorno, llegar al diagnóstico clínico y molecular puede ser un reto importante para los profesionales de la salud. Presentamos el primer caso descrito de un paciente con AT en la República Dominicana.
Presentamos el estudio de caso clínico de un niño de 7 años, de ascendencia dominicana; hijo único de padres no consanguíneos. Antecedente materno de trastorno hipertensivo durante el tercer trimestre de la gestación, uso regular de vitaminas prenatales, teratógenos negados. Además, no se constatan otros antecedentes familiares relevantes.
En cuanto a los antecedentes personales, el paciente mostró un retraso del crecimiento intrauterino como hallazgo sonográfico a partir del quinto mes de embarazo. Al nacer, se objetiva como un niño eutrófico, vía cesárea, G1/1, a término, peso: 5 5/16 libras, APGAR 8/9, en alojamiento conjunto a la madre. Además, presentó infecciones respiratorias acompañadas de crisis de sibilancias recurrentes y abscesos múltiples en piel durante la infancia.
A partir de los 6 meses de edad inició con dificultades para adquirir los hitos del desarrollo motor, con posterior inestabilidad postural al sentarse y pararse; además de una marcha en puntillas y atáxica.
Fue progresando con una neuropatía motora periférica que se evidenciaba como limitaciones para ejecutar actividades cotidianas como subir escaleras, correr, hacer deportes, entre otros. Posteriormente, en su evolución, presentaba disfagia y disartria. Además, el paciente manifestaba infecciones respiratorias recurrentes y abscesos múltiples, así como lesiones eritematosas en la piel.
Por otro lado, en cuanto a la exploración física (véanse figuras 1-3), se evidencia retraso pondoestatural, deterioro neurológico, diámetro bifrontal ligeramente estrecho, hipertelorismo, epicanto, telangiectasias oculares en ambos ojos, sialorrea y disartria; cardiovascular normal, con estertores roncus bilaterales en ambos campos pulmonares, telangiectasias en ambas palmas de las manos, máculas hipo e hipercrómicas residuales en piel generalizados, marcha atáxica, con ligera rotación de los pies, fuerza muscular disminuida (4/5) y sensibilidad conservada.
Fuente: expediente Clínico. Gen & Kids Center.
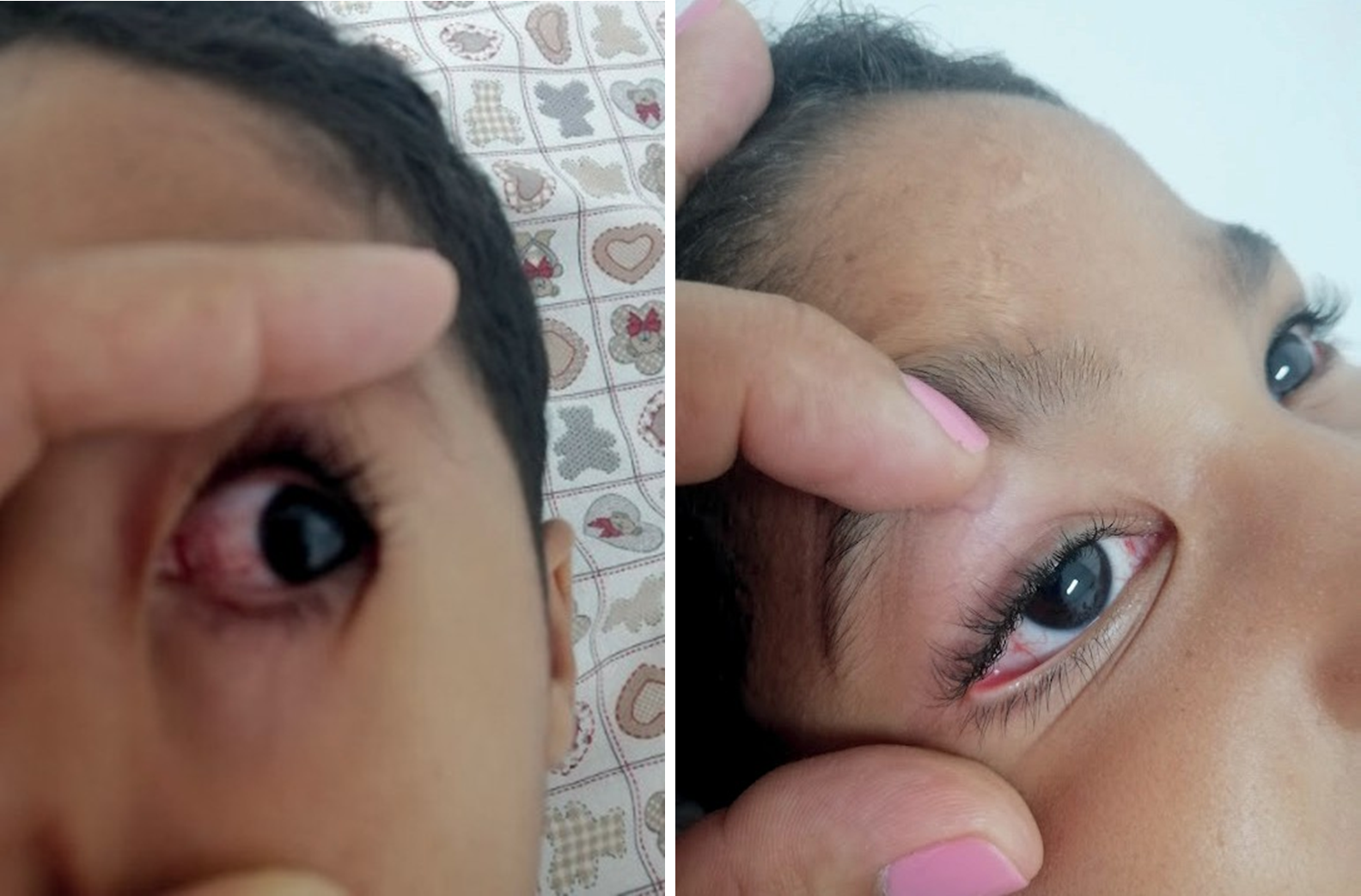
En las figuras 1 y 2 se evidencian telangiectasias oculares.

Figura 3. Telangiectasias en palmas de las manos
Fuente: expediente Clínico. Gen & Kids Center.
Las pruebas complementarias realizadas incluyeron un hemograma, perfil hepático, renal, tiroideo, creatina quinasa total, los cuales resultaron normales, y niveles de alfafetoproteína elevados según parámetros de referencia de laboratorio. Igualmente, fue realizada una evaluación cardiovascular, una electromiografía y un electroencefalograma sin hallazgos. En cambio, la resonancia magnética cerebral mostró una atrofia cerebelar.
Tras la recogida de antecedentes personales y familiares, el examen físico y análisis complementarios se identifica como diagnóstico clínico ataxia-telangiectasia. Por tanto, fue llevada a cabo una prueba genética molecular en sangre periférica consistente en la secuenciación del gen ATM con el objetivo de confirmar diagnóstico presuntivo, obteniéndose un reporte positivo con la identificación de una variante genética en homocigosis, ATM: c.7913G>A (p. Trp2638*), clasificada como patogénica.

Figura 4. Prueba genética molecular (secuenciación gen ATM). Variante patogénica en homocigosis asociado con diagnóstico de ataxia-telangiectasia autosómica recesiva.
Fuente: Expediente Clínico. Gen & Kids Center.
El paciente recibe tratamiento multidisciplinario con genética, neurología pediátrica, oftalmología, inmunología, neumología, endocrinología, ortopedia, vigilancia oncológica, terapia física, ocupacional y del habla.
El estudio de caso presentado aborda la complejidad de la AT, una enfermedad neurodegenerativa y multisistémica con una amplia heterogeneidad clínica, abarcando desde formas clásicas con fenotipos más severos hasta manifestaciones más leves. Pertenece al grupo de trastornos hereditarios conocidos como síndrome de inestabilidad cromosómica junto al síndrome de Bloom, la anemia de Fanconi y el síndrome de Nijmegen; asociados con inmunodeficiencias y riesgo aumentado de neoplasias malignas.
El paciente descrito, presenta una combinación de síntomas neurológicos, como ataxia cerebelosa progresiva, disartria y neuropatía motora, junto con manifestaciones sistémicas, como infecciones respiratorias recurrentes, abscesos cutáneos, telangiectasias y retraso de crecimiento. Además de la identificación de atrofia cerebelar en la resonancia magnética craneal, lo que sustenta la sospecha diagnóstica de AT.
Las pruebas complementarias, incluyendo los test genéticos, son fundamentales para confirmar el diagnóstico de AT y para identificar las variantes patogénicas específicas en el gen ATM. En el caso de nuestro paciente, la variante identificada ( ATM c.7913G>A (p.Trp2638X) está clasificada como patogénica y ha sido descrita anteriormente en diversas publicaciones científicas7-10. Esta variante resulta en un codón de terminación prematuro, el cual resulta en una proteína ausente o no funcional9. Igualmente, el estado de homocigosidad corrobora el diagnóstico de este caso.
Destacamos la importancia del asesoramiento genético en el manejo de la AT, especialmente en la identificación de portadores y en la evaluación del riesgo oncológico en familiares. Dada la asociación de la AT con un aumento significativo en el riesgo de malignidad, se enfatiza la necesidad de una vigilancia y seguimiento adecuados, especialmente en mujeres portadoras.
En resumen, este es el primer estudio de un caso publicado de ataxia-telangiectasia en la República Dominicana. Este proporciona una visión detallada de la presentación clínica de la AT, resaltando la complejidad diagnóstica y la importancia del enfoque multidisciplinario para abordar los aspectos neurológicos y sistémicos de esta enfermedad rara.
En esta publicación científica se han seguido los criterios éticos estándares para casos clínicos patológicos, preservando los datos del paciente en anonimato, con previa autorización bajo consentimiento informado para toma y manejo de las imágenes. Respetando los lineamientos y normativas establecidas para la redacción y formato de esta.
La presente investigación científica no ha recibido ningún tipo de financiamiento para llevarse a cabo.
Las autoras del artículo declaran no poseen conflictos de intereses para la publicación de este.